The fundamental ethic of the well-characterized biopharmaceutical is that drug developers can define and monitor the critical attributes of a biotherapeutic, and that they can produce a process that maintains control over these characteristics. The faster you can define the underlying structural variation in a product, the faster you can proceed through product commercialization and implement process improvements.
Peptide mapping by LC-MS is an essential technique used by the biopharmaceutical industry to examine the primary structure of a protein. Mass spectrometry provides an extra dimension of separation over traditional UV detection methods. This removes the absolute reliance on effective chromatographic resolution to enable comprehensive qualitative and quantitative peptide map analysis. In an LC-MS peptide map, peptides can partially or fully coelute without impairing the ability to detect, assign, and quantify them. Thus, an MS detection workflow enables product characterization to proceed independently of the time-consuming efforts to develop the more resolving LC-UV peptide maps that are required for later development and quality control activities. The challenge with implementing LC-MS peptide mapping has been that data processing and interpretation were productivity-limiting.
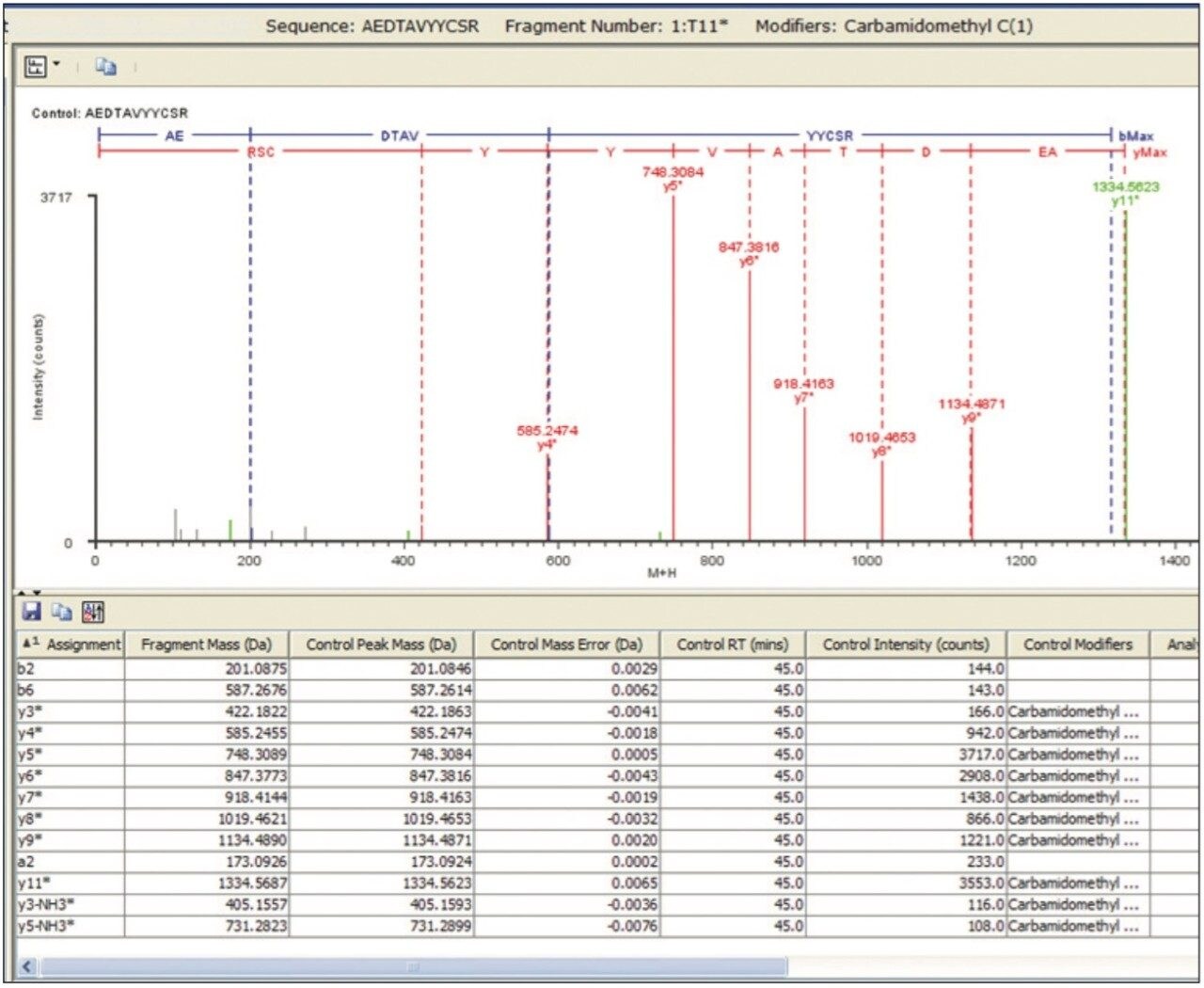
The introduction of data-independent acquisition approaches such as UPLC-MSE has enabled shorter peptide mapping runs, and simultaneously increased the amount and quality of information available for biotherapeutic characterization. MSE is a technique that acquires accurate peptide and fragment mass data for all peptides within a sample within a single run. The MSE fragmentation data can be used to validate the accurate mass assignment of a peptide and localize assigned modifications within the peptide sequence. Fragmentation information is collected in parallel for all peptide ions, avoiding the bias and analytical irreproducibility that occurs with data-dependent approaches that require serial pre-selection of peptide precursor ions. LC-MSE datasets are acquired with two alternating MS functions: one for MS of peptide precursors acquired at low collision cell energy, and one (MSE) for collecting peptide fragmentation data at elevated collision cell energies.
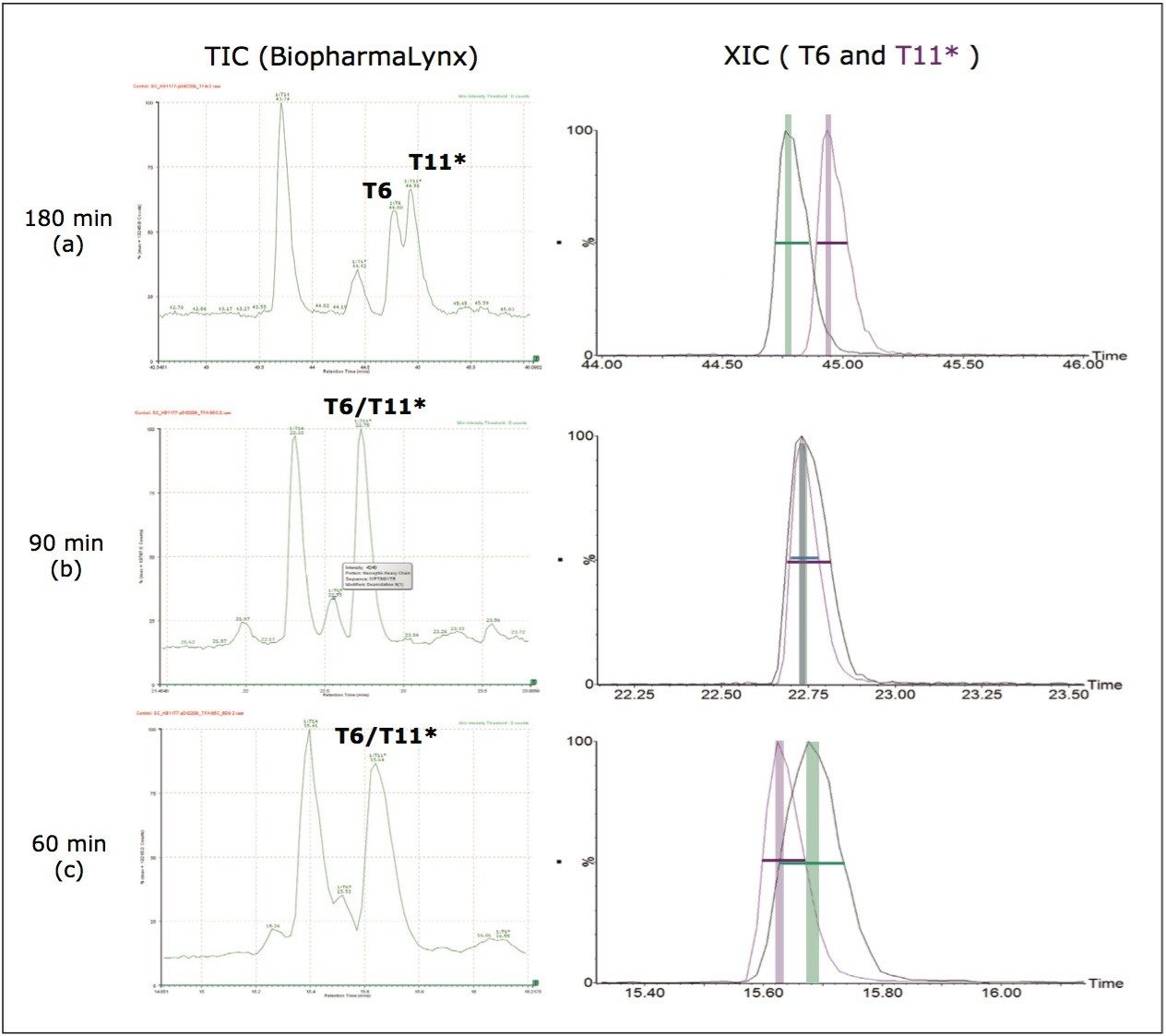
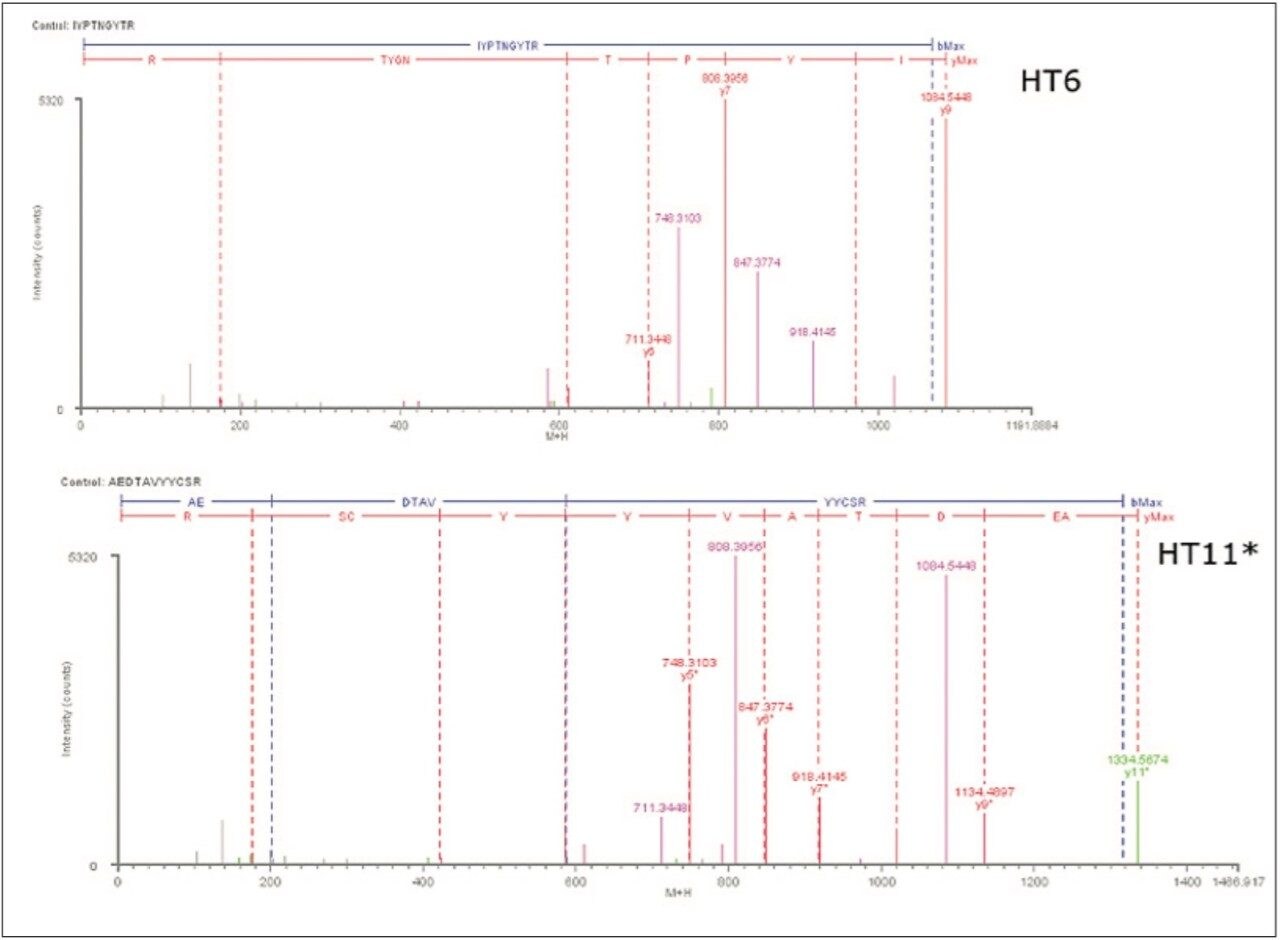
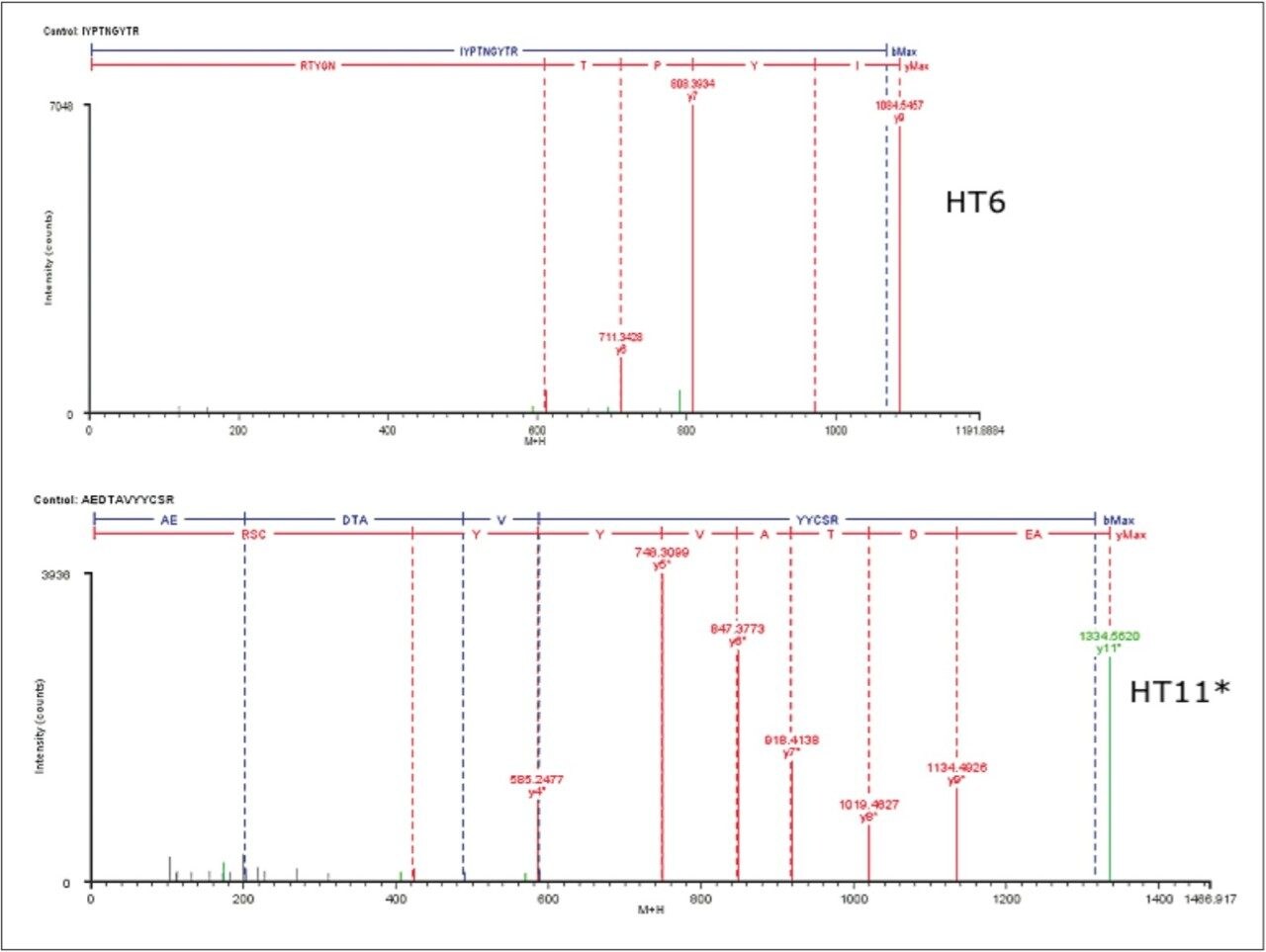
Several publications1-5 have detailed the power of processing such datasets using the chromatographic profiles of detected ions to determine isotopic cluster, charge state, and precursor ion/fragment ion relationships in MSE datasets. Using this methodology, the resulting peptide information included retention time, accurate mass, intensity, and fragmentation profiles for all detected peptides within a peptide map.
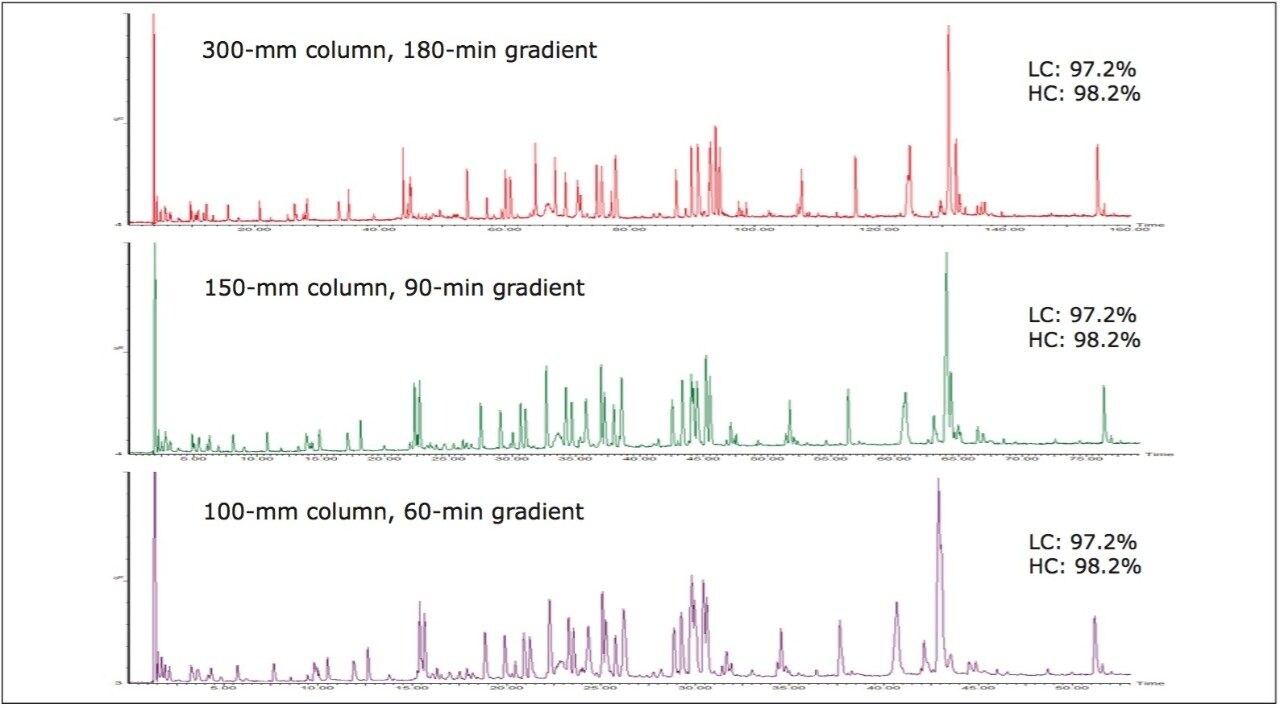
In this application note, we show how BiopharmaLynx software quickly automates assignment of peptide sequence information from MSE fragmentation data, even for cases of coeluting peptides. The ability of the LC-MSE approach to reduce mAb peptide mapping run times from 180 min (300-mm column) to 60 minutes (100-mm column) without affecting peptide sequence coverage and fragment pattern interpretation is demonstrated.
Automating data analysis enables analysts to realize the increased productivity gained from using shorter LC-MS methods for product characterization studies. Furthermore, the ability of the underlying MSE methodology to deal with high-complexity data enables the production of generic LC-MS screening methods for a wide array of biotherapeutic products. Productivity gains on the order of weeks to months can be expected compared to time spent optimizing and analyzing LC-UV peptide maps.