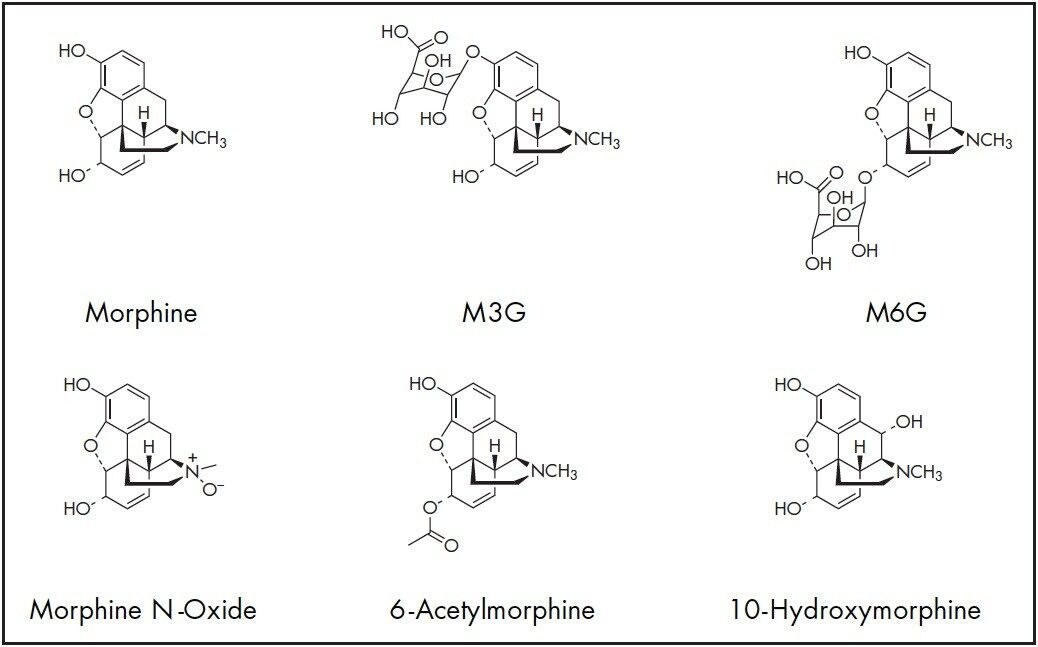
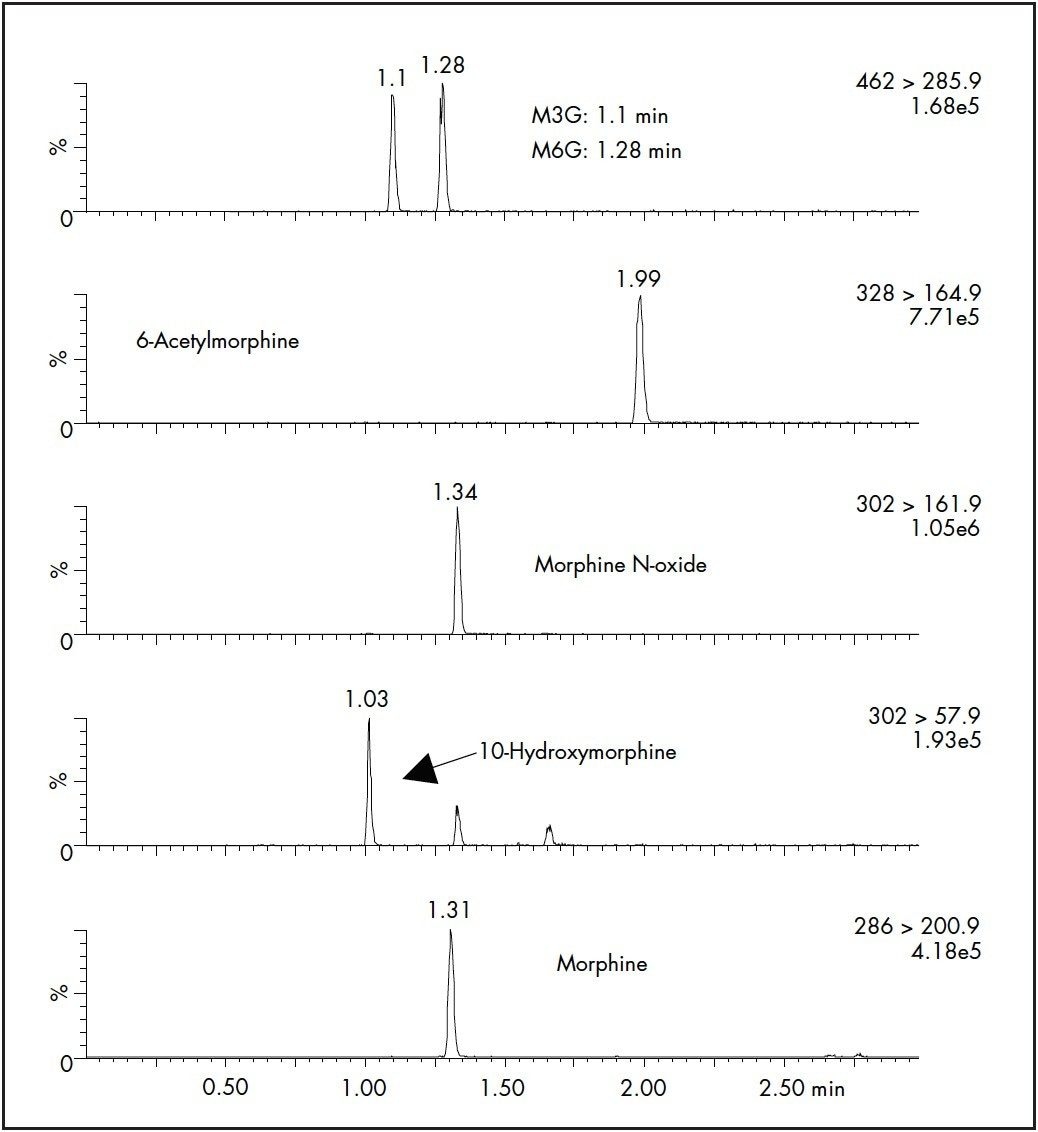
Morphine is an effective pain-relieving drug that is primarily metabolized into morphine-3-glucuronide (M3G) and morphine- 6-glucuronide (M6G). The highly potent M6G may have adverse effects such as respiratory depression and renal failure if accumulated in the body. Other metabolites of morphine include morphine N-oxide, 6-acetylmorphine and 10-hydroxymorphine. As morphine abuse continues to affect modern society, an effective method must be established to analyze morphine and its metabolites in biological fluid samples.1
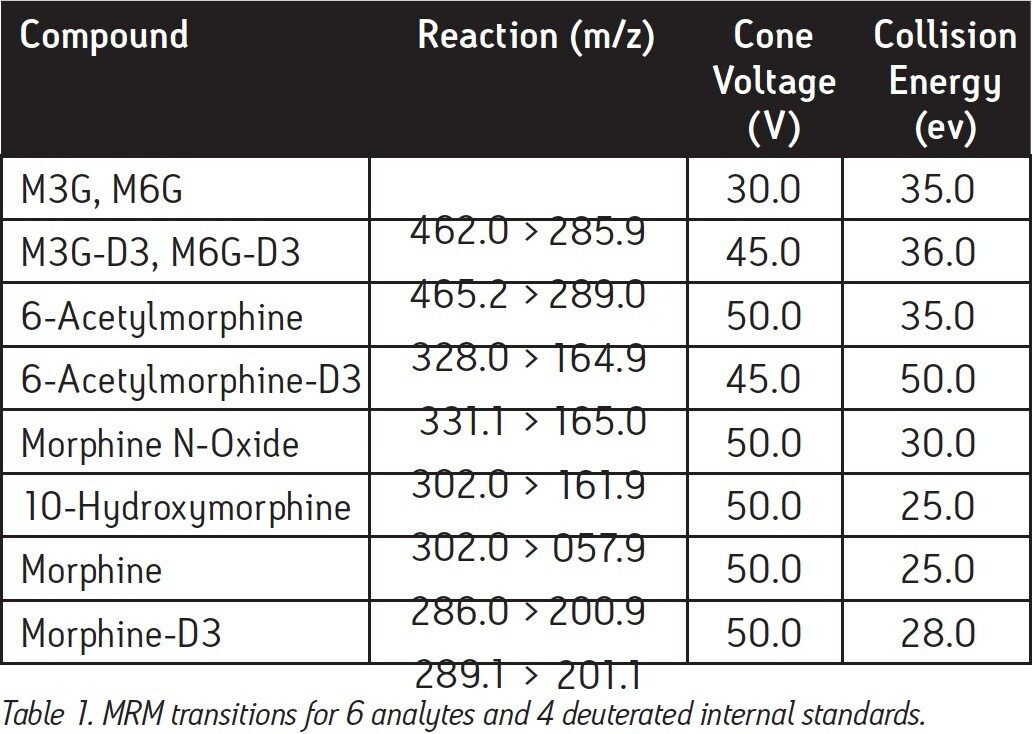
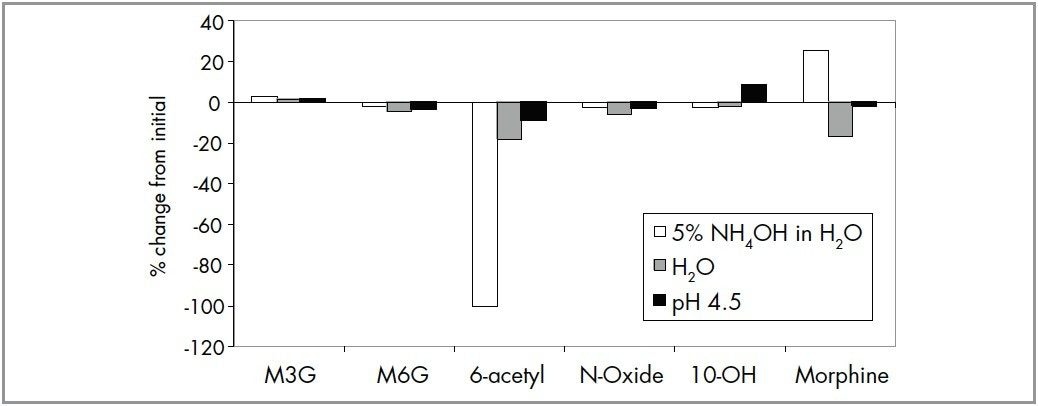
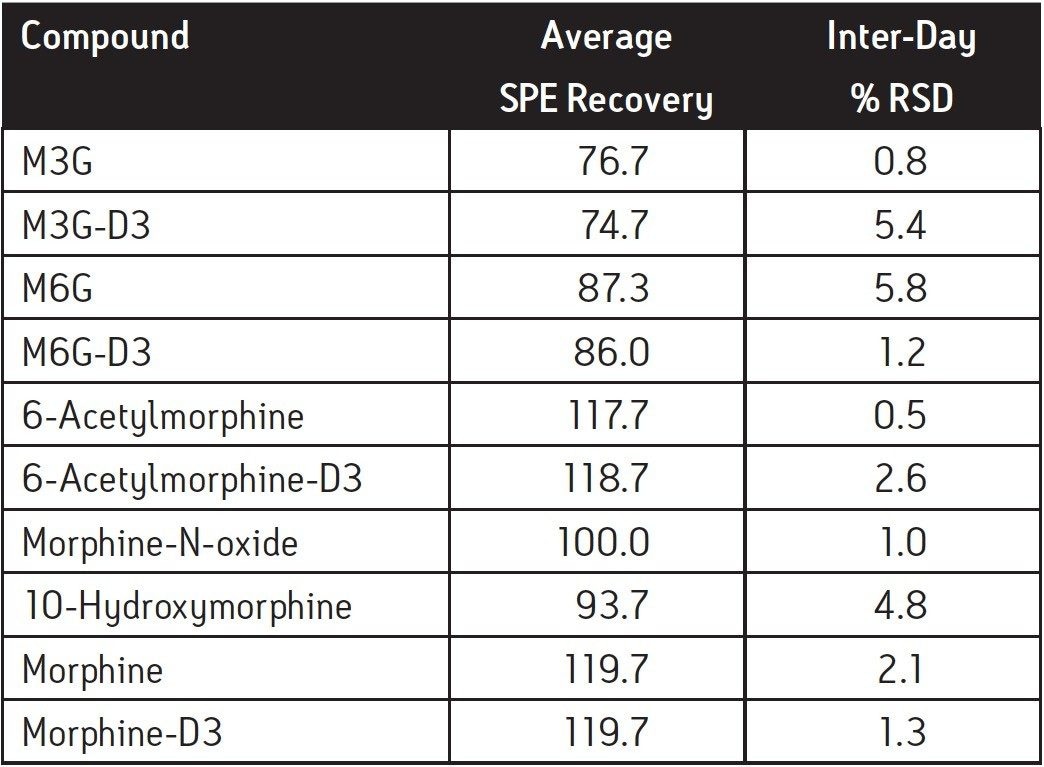
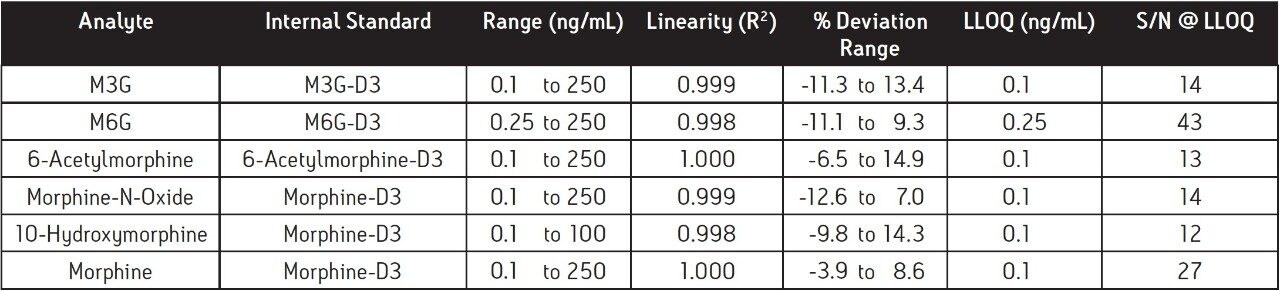
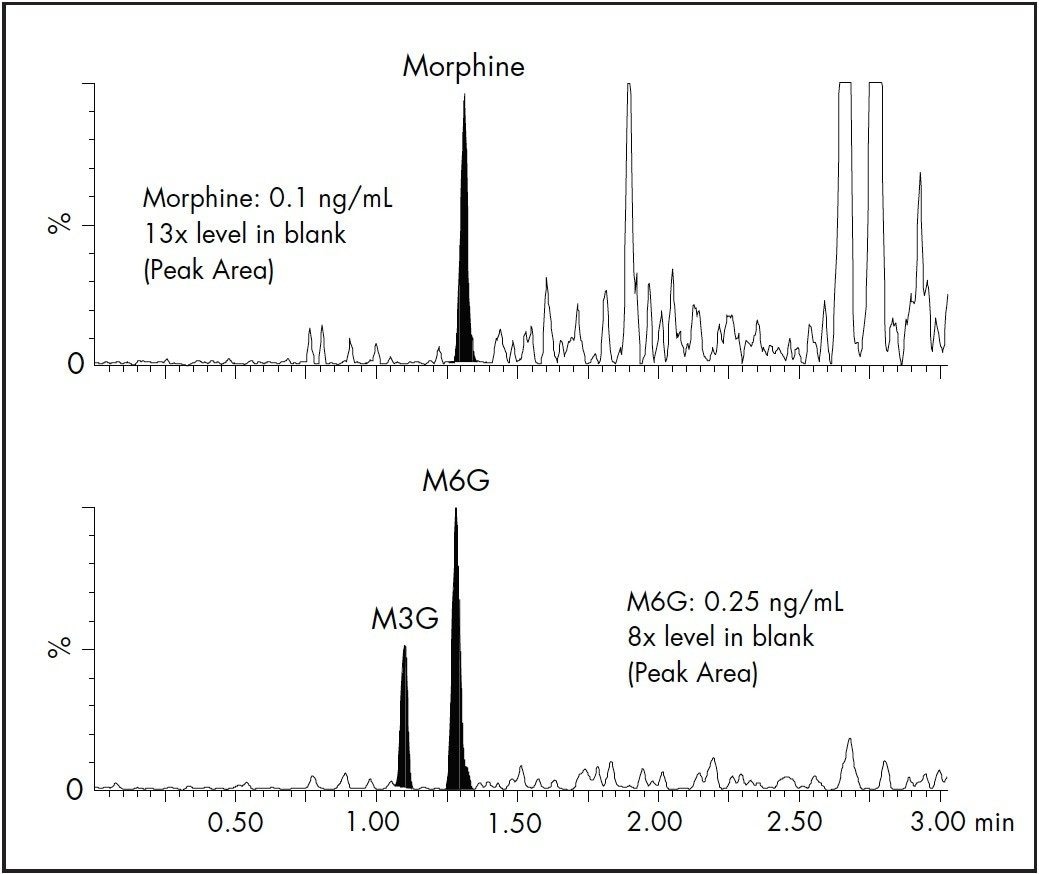
In this work, a UPLC-MS/MS method was developed to separate morphine and five of its metabolites on a 2.1 x 100 mm, 1.8 μm ACQUITY UPLC HSS T3 Column in a single run using an ACQUITY UPLC System connected to a fast-scanning triplequadrupole MS detector (TQD). The method achieved adequate retention of these very polar compounds by reversed-phase (RP) chromatography in an 8-minute total run time. Mixed-mode solid-phase extraction (SPE) uses both reversedphase and ion-exchange mechanisms to separate analytes more selectively from matrix components. Therefore, sample preparation of porcine plasma was performed with the Oasis Mixed-mode Cation eXchange (MCX) μElution Plate. The strong cation-exchange sorbent was chosen because morphine, its five metabolites and, their internal standards are basic compounds (pKa of morphine = 9.85). The structures of the six analytes are shown in Figure 1. The Oasis MCX SPE procedure requires a high-pH elution step that is not suitable for 6-acetylmorphine due to degradation. To avoid compound degradation prior to UPLC-MS/MS analysis, a neutralizing collection step was employed.2 The SPE recovery and reproducibility of the method were determined, as well as the linearity and lower limit of quantitation (LLOQ) for each analyte.