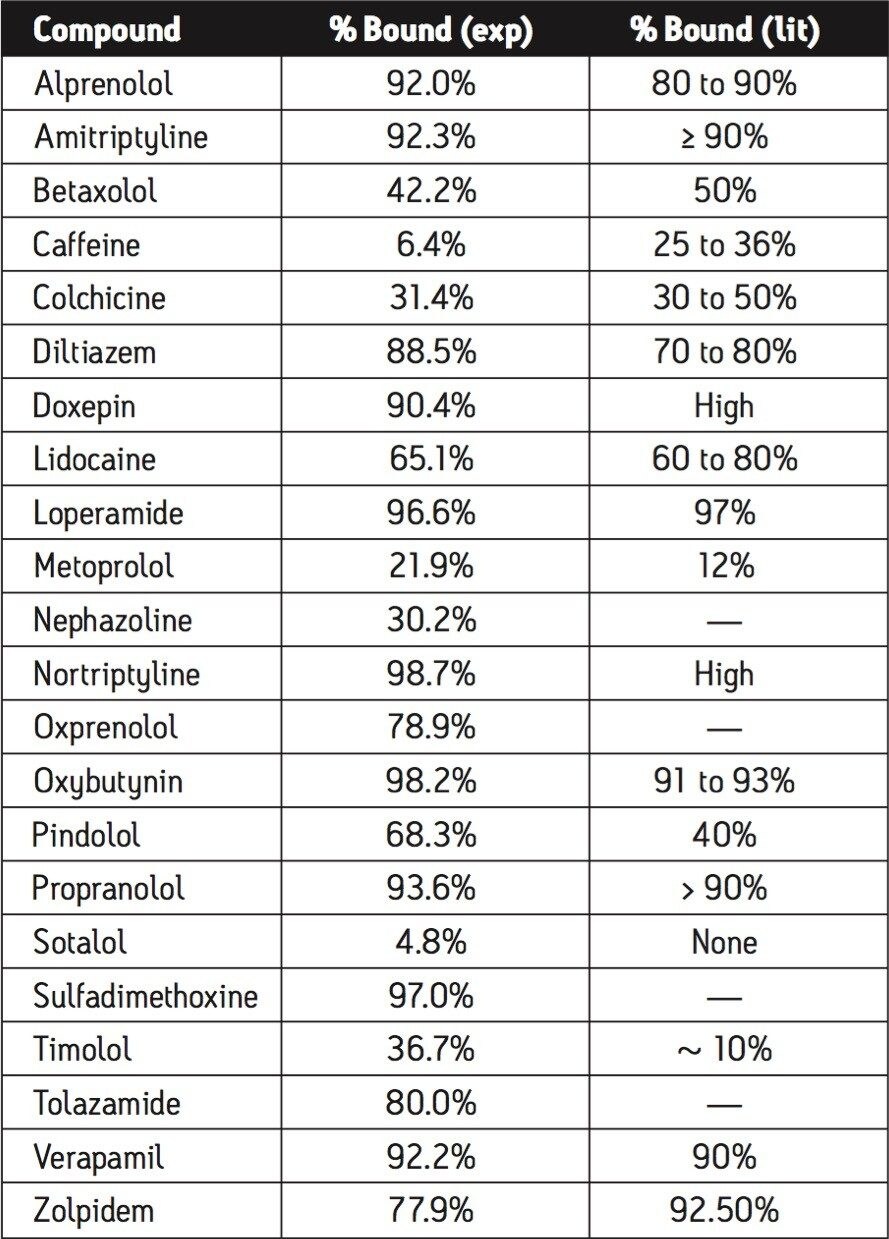
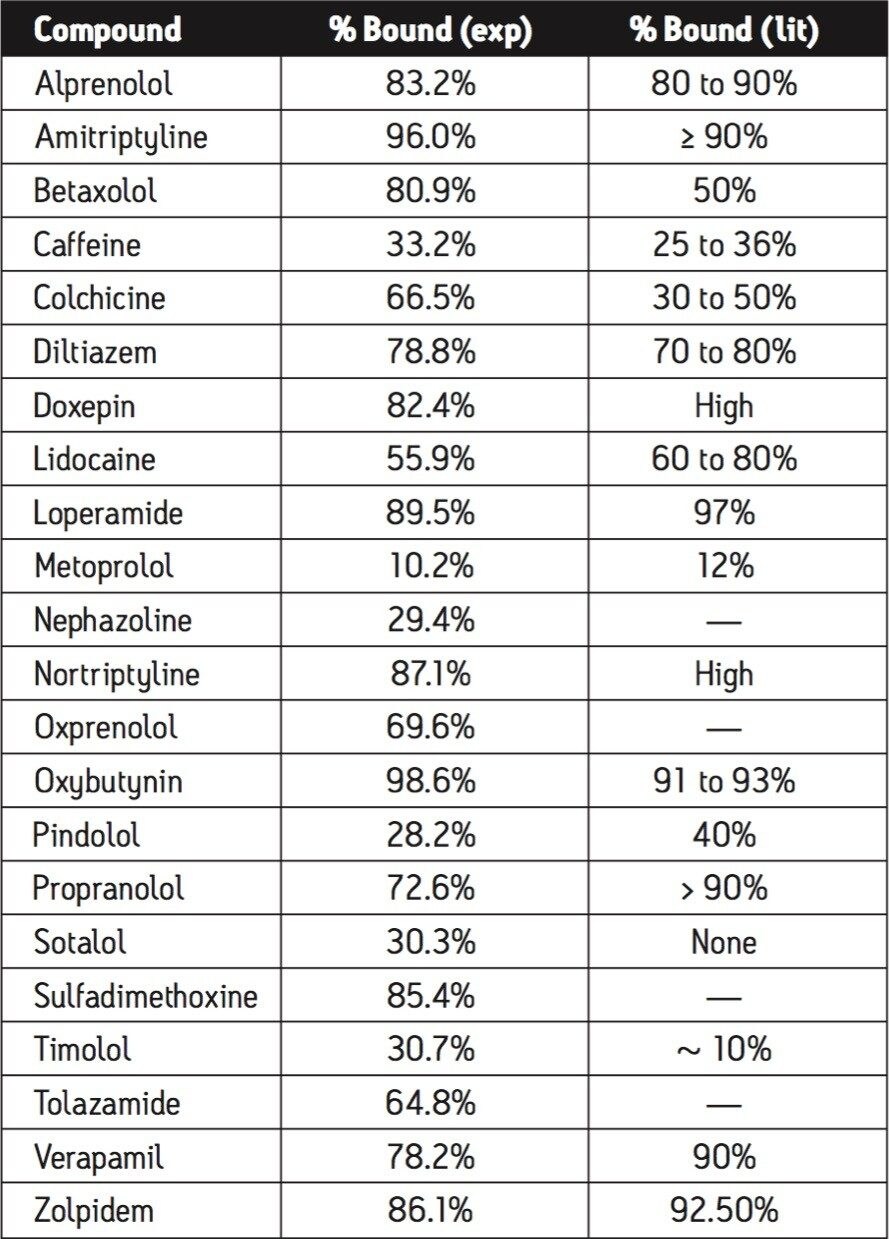
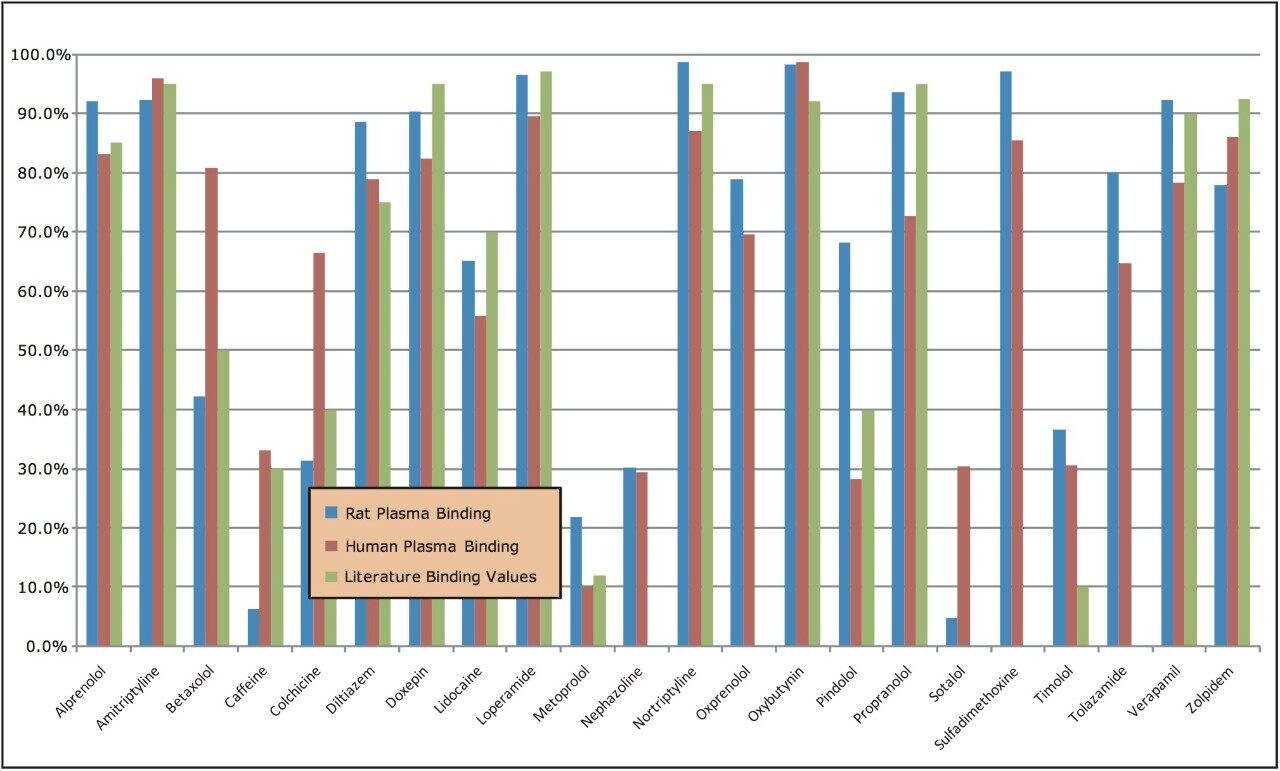
Plasma protein binding (PPB) can significantly affect the therapeutic action of a drug. PPB determines the extent and duration of action, as only unbound drug is thought to be available for passive diffusion to extravascular or tissue sites where therapeutic effects occur.
Since data show an increasing importance of PPB in characterizing a drug’s behavior and proper dosing, PPB measurement needs to be made as early as possible in the discovery process. In vivo dose levels can be estimated from the determined fraction of unbound drug (fu); a drug that demonstrates high plasma protein binding indicates that an increase in dose might be necessary.
The classical method used to measure the level of protein binding is equilibrium dialysis. In equilibrium dialysis, a dialysis membrane with small pores allows molecules to diffuse through it. Once equilibrium has been reached, one can measure the amount of free analyte in the donor and receptor samples, and then determine the amount of bound analyte.
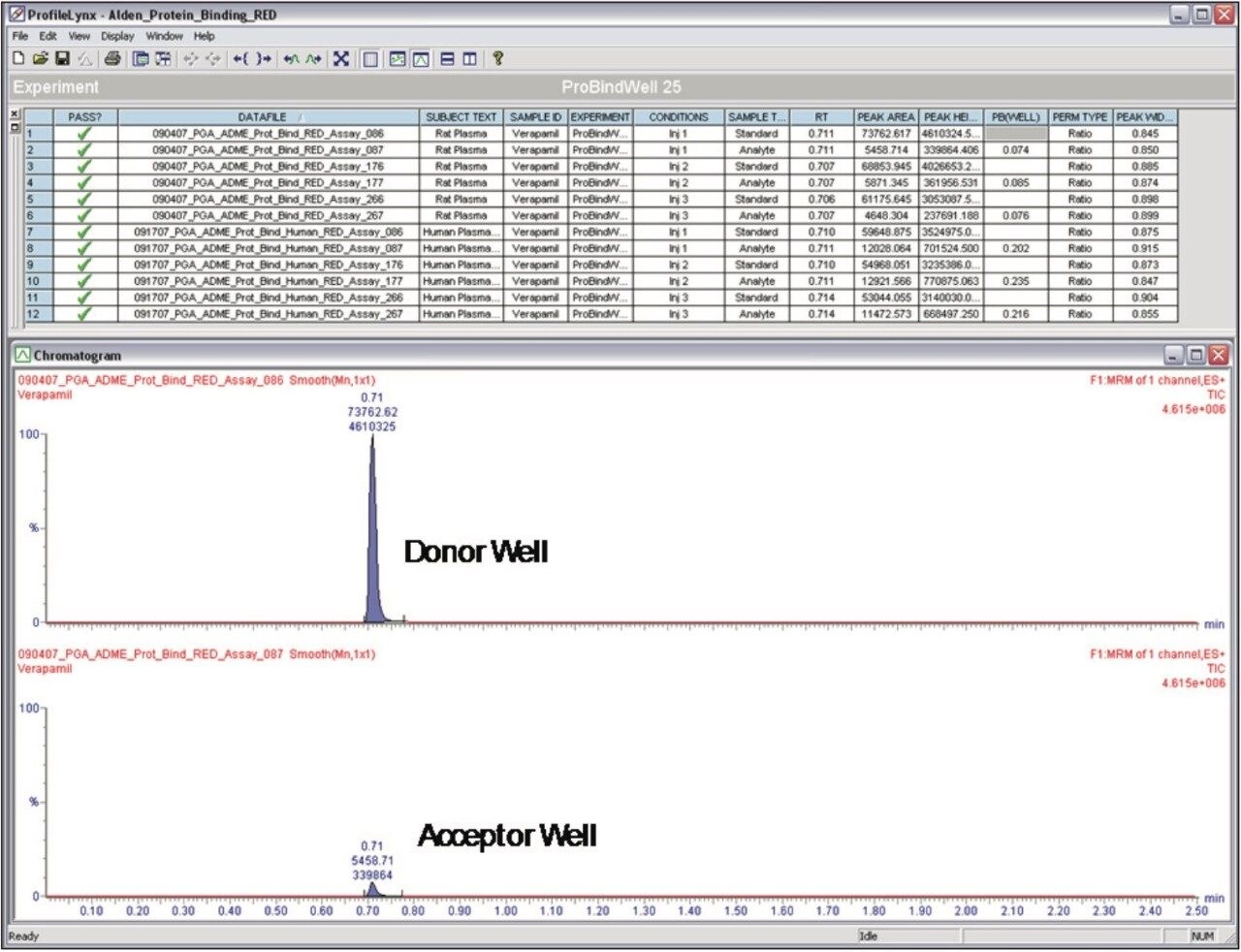
This process is laborious and time-consuming with the need to perform additional analytical steps, including radiolabeling. Given the detection speed and sensitivity of UPLC-MS/MS, the ACQUITY TQD System (Figure 1), used with specialized software, ProfileLynx and QuanOptimize Application Managers, is the ideal choice for analysis of PPB.