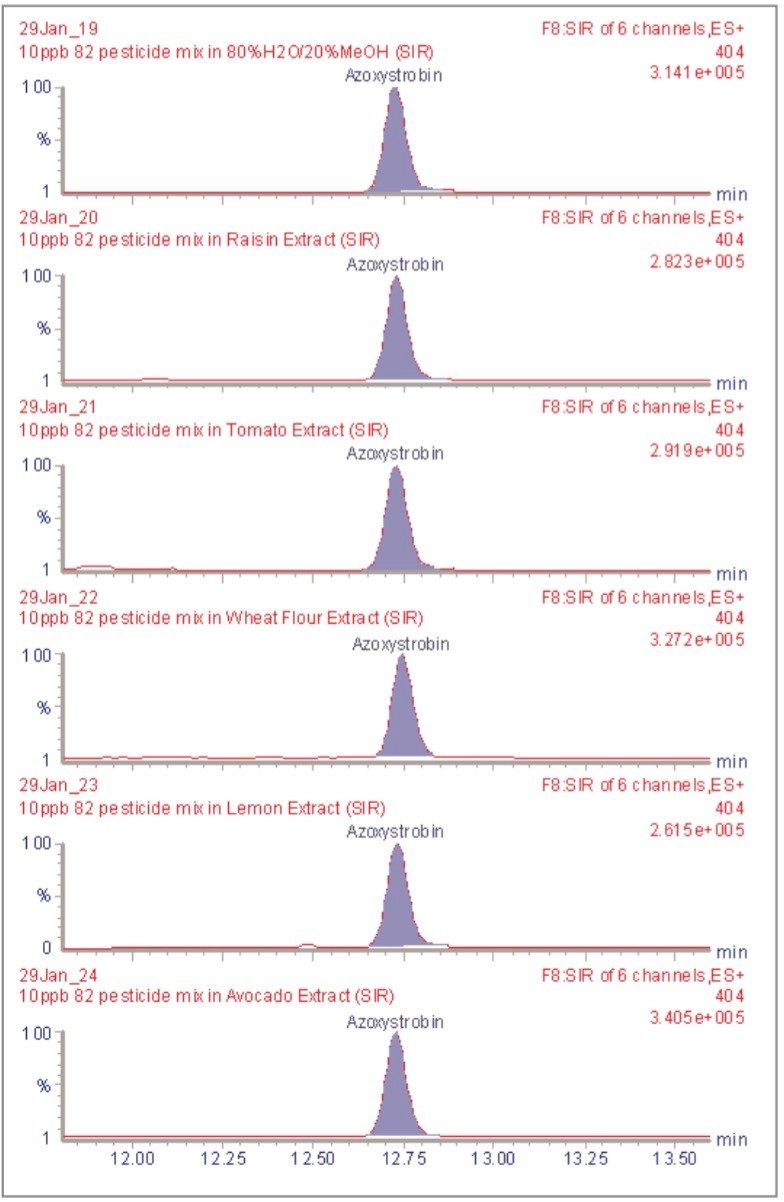
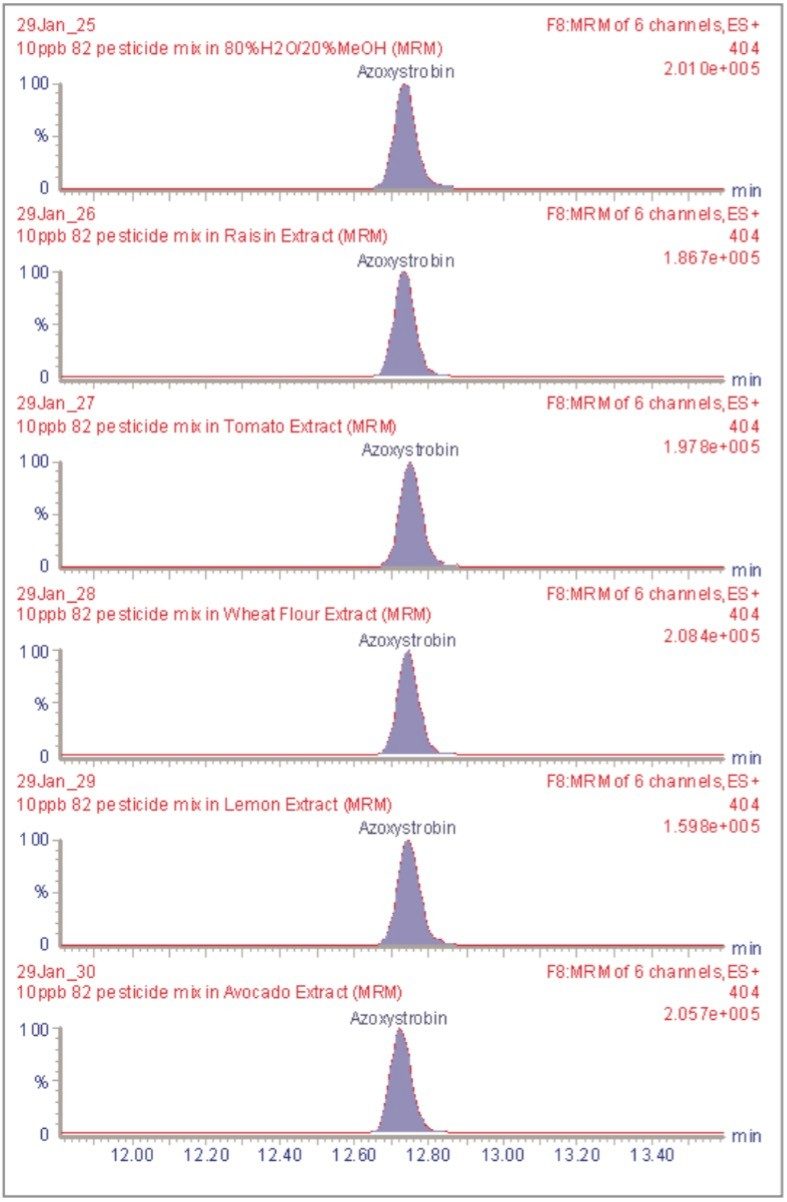
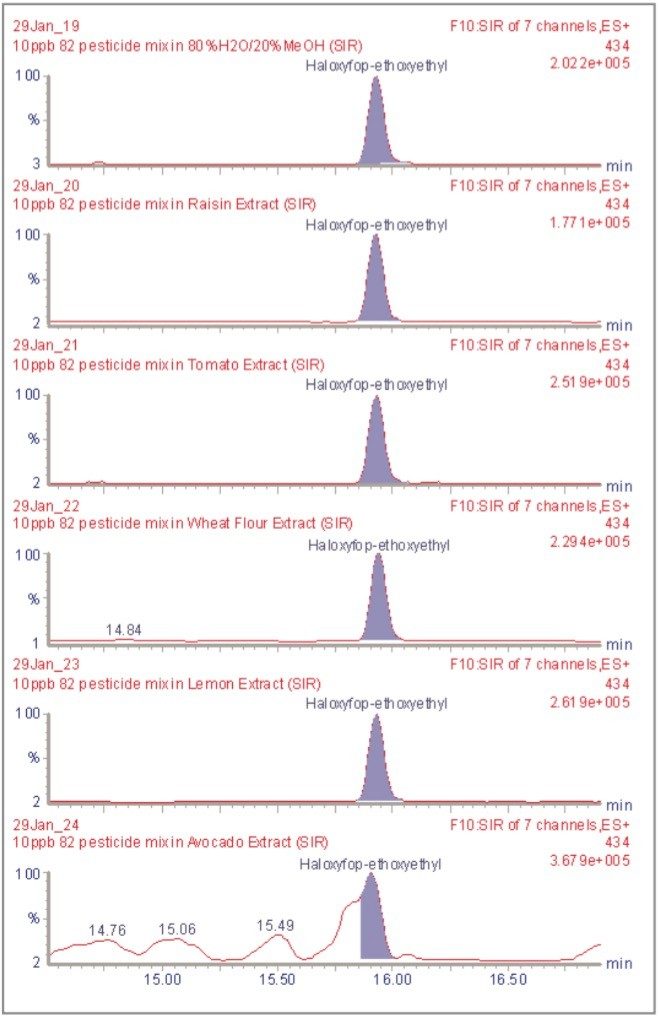
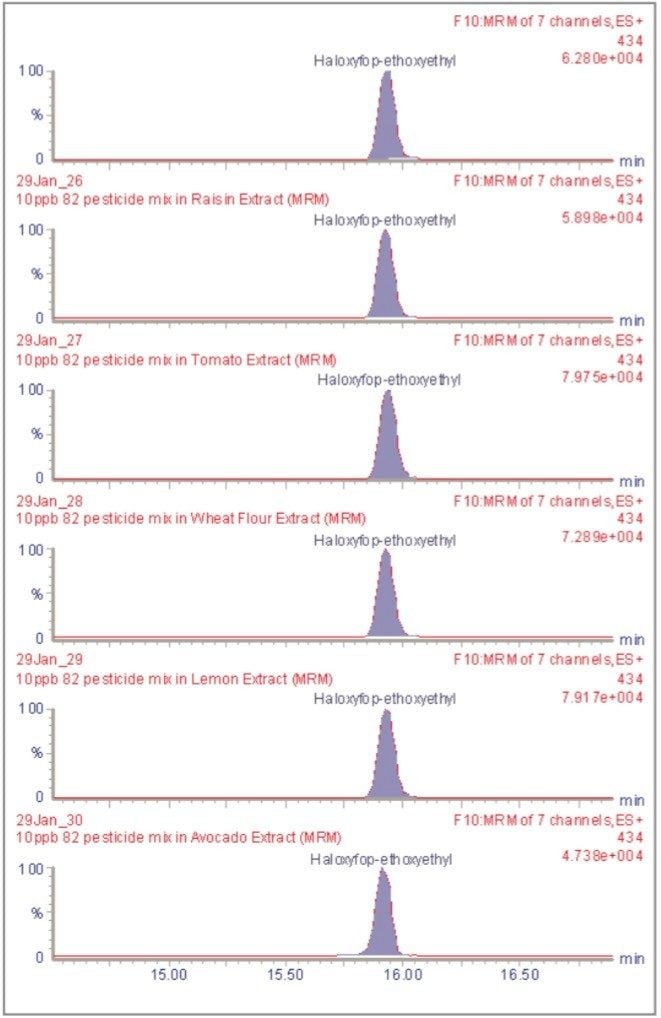
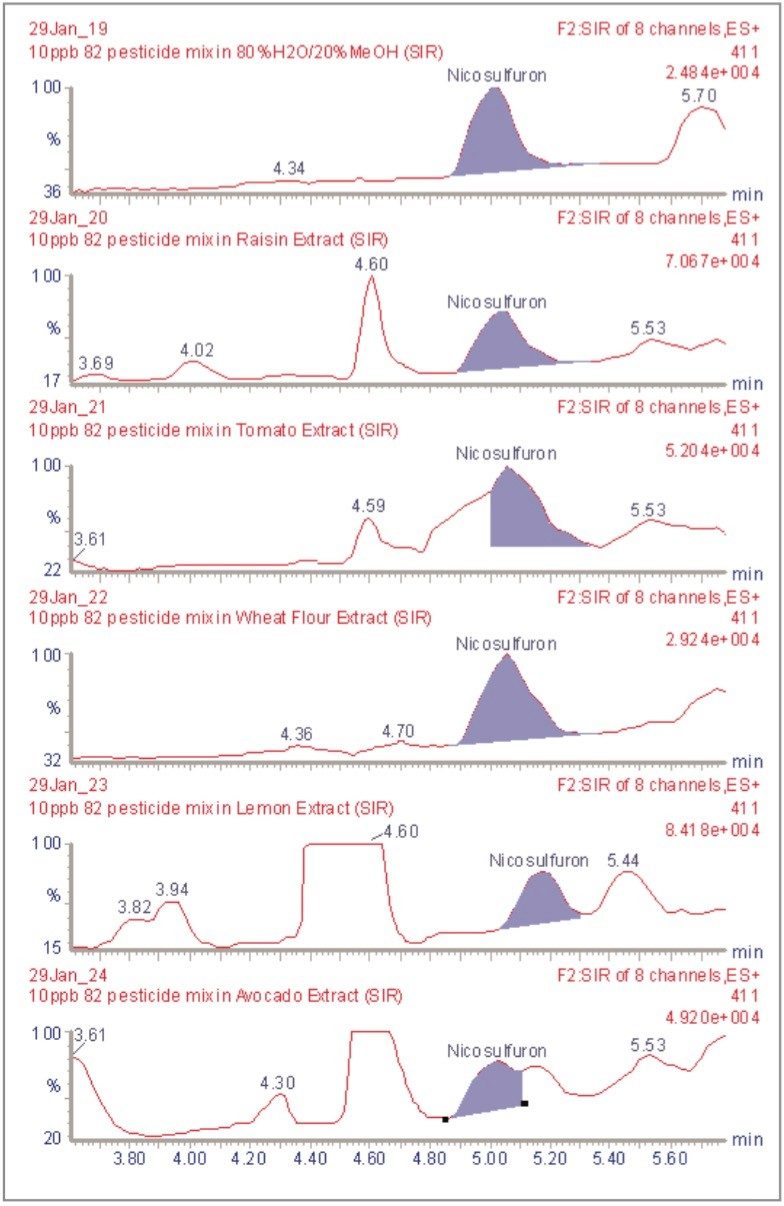
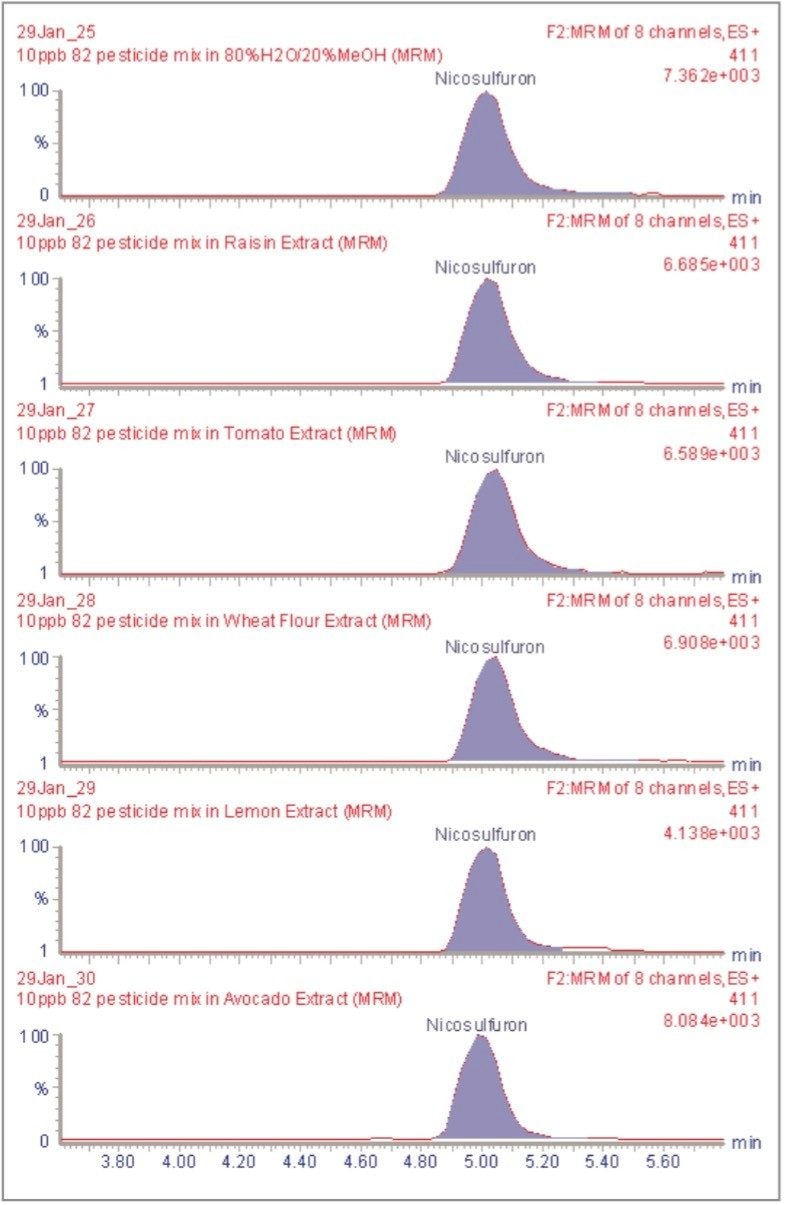
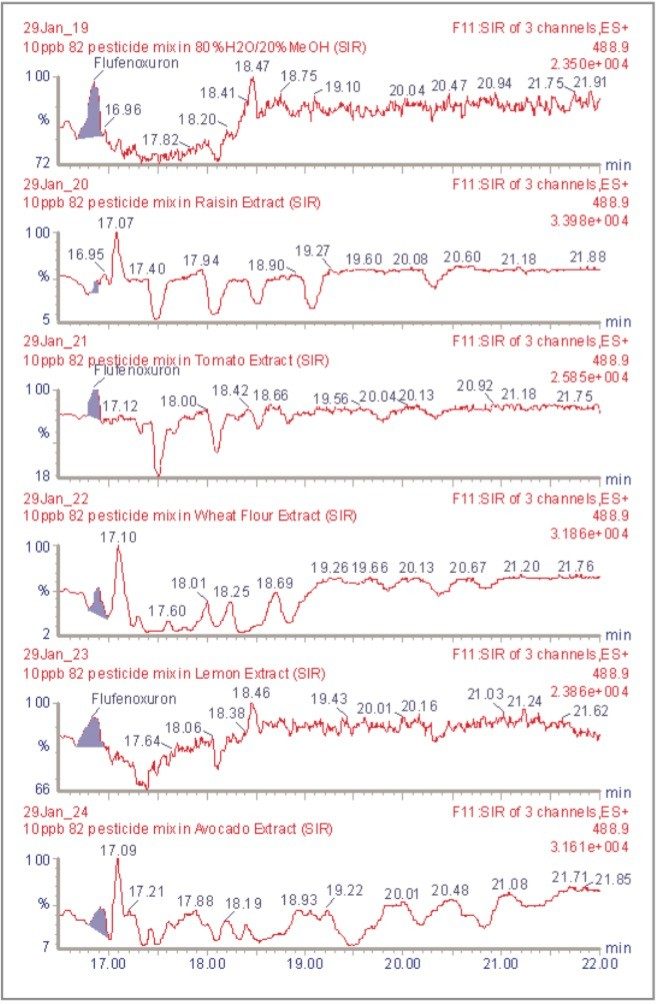
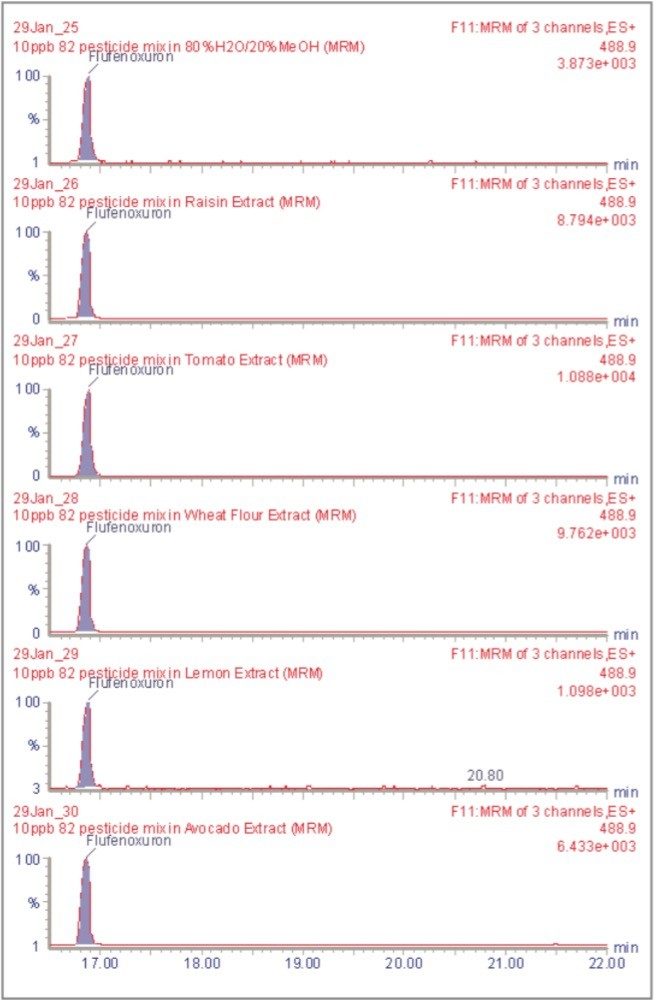
The inappropriate or unlawful use of pesticides on agricultural crops can result in unacceptably high levels of these compounds, and their metabolites, in produce destined for human consumption. To protect the health of consumers, many countries stipulate Maximum Residue Levels (MRLs) for each pesticide compound in a range of fruit and vegetables. Worldwide, there are over 800 compounds currently in use to control pests such as insects, weeds, rodents and fungi. The legal enforcement of regulations governing pesticide use requires the regular monitoring of agricultural produce. Given the large number of pesticide residues that may be found, it is advantageous to determine as many of them as possible during a single analysis multi-residue methods have been developed that target more than one analyte compound. As the number and diversity of target analytes is increased, the selectivity of the clean-up stage of sample preparation is necessarily compromised, resulting in a more complex sample matrix.
To maximise the efficient use of analytical resource it is also advantageous to have a general sample preparation method that is applicable to a variety of produce. Such a method will produce sample matrices of various compositions, depending on the type of produce under investigation. The potential for analytical interference from co-extracted substances is high, and the analytical selectivity of such a multiresidue, multi-produce method must, therefore, be provided by the determinative step. Mass spectrometry is a highly selective analytical technique that can be used to monitor specific ions generated from the analytes of interest. The use of the Selected Ion Recording (SIR) method provides a greater level of selectivity than other detection methods such as UV/Vis spectrometry. However, when the analysis of 81 pesticide residues is required, the low selectivity of the clean-up stage means that the SIR method does not eliminate the potential for interference from matrix components.