Curation of MRM Transition Candidates in Method Development for Quantification of Peptides in Protein Digest Samples
Este é um Resumo de aplicações e, por isso, não inclui uma seção de experimento detalhada.
Abstract
To detect and quantify peptides representing target proteins in complex biological samples, while avoiding certain analytical situations that complicate the analysis, and may produce erroneous results.
Benefits
Learn the importance of peptide selection for quantification studies using tandem quadrupole MRM analysis.
Introduction
A companion technology brief1 describes the excellent quantitative results for MRM analysis of peptides in protein digests when all of the following conditions are met:
1. The peptide(s) chosen for analysis is/are unique to the protein of interest.
2. The peptide ionizes with sufficient efficiency to attain the desired detection limit.
3. No other peptides with precursor and product masses similar enough to those of the peptide of interest to produce signal in the MRM data channel elute at retention time similar enough to the peptide of interest to interfere.
4. The peptide exhibits good chromatographic peak shape.
When conditions 3 and/or 4 are not met, it will be difficult or impossible to obtain satisfactory quantitative results, as will be illustrated in this technology brief.
Results and Discussion
The Solution
Five tryptic peptides for each of the four proteins comprising MassPREP™ Protein Expression Mixture 1 Digestion Standard were selected for MRM analysis, and several MRM transitions were chosen for each peptide. Mixture 1 was analyzed using a Xevo® TQ Mass Spectrometer with a nanoACQUITY UPLC® System equipped with a 75 μm diameter column using 1 μL injections. The mixture was injected at 100 fmol/μL by itself, and at concentrations ranging from 200 fmol/μL to 390 amole/μL in a matrix of 1 μg/μL of digested human serum proteins.
The results were curated according to the criteria listed above. Here, we describe some examples which illustrate conditions 3 and 4.
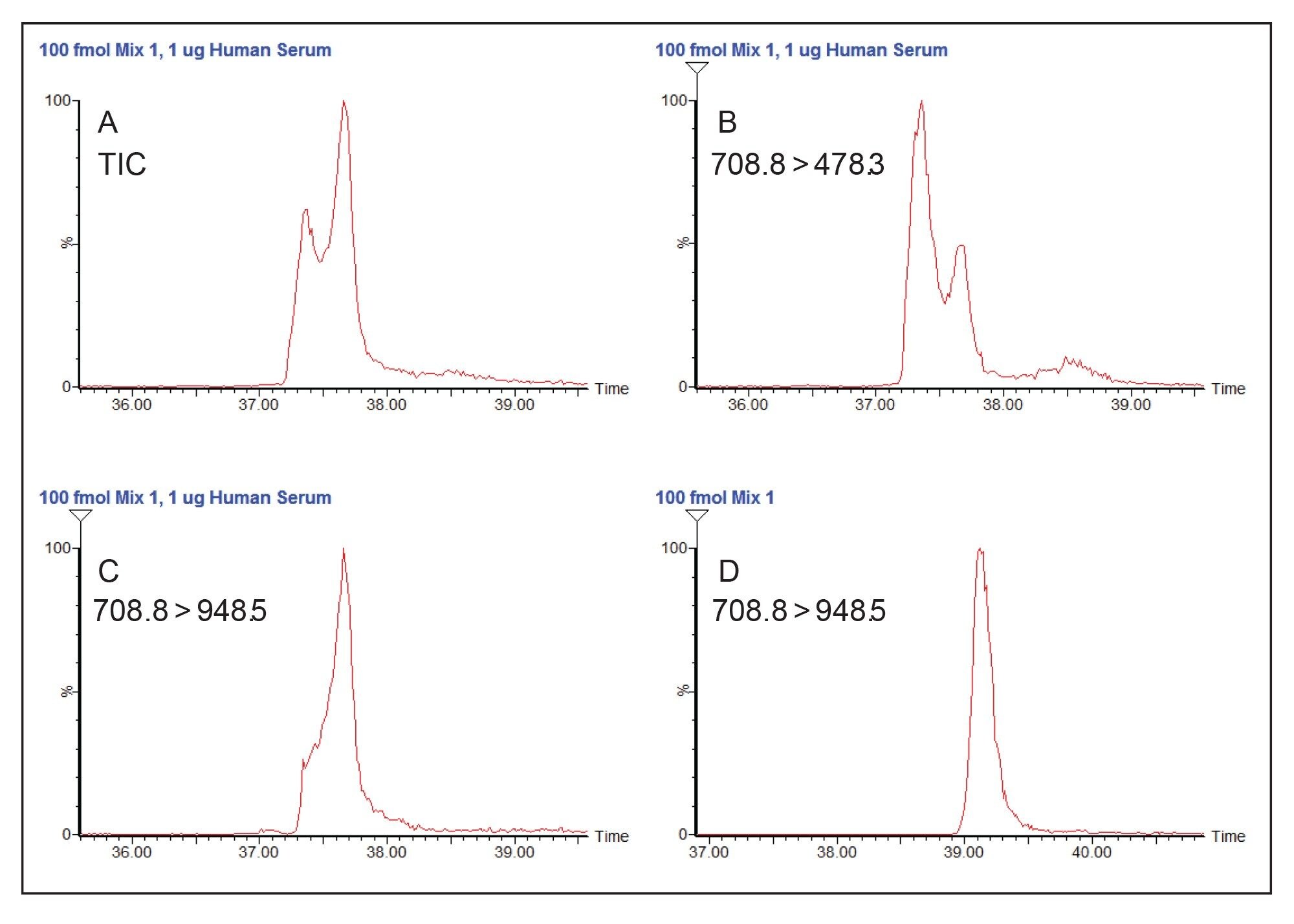
Figure 1A shows the TIC chromatogram for the yeast enolase peptide GNPTVELTTEK, where the analyte peak on the right overlaps with that of an interfering peptide on the left. Much of the intensity of the interfering peptide is contributed by the 708.8 to 478.3 transition (Figure 1B), but this peptide produces signal in other transitions as well, as seen by the distorted peak shape in 1C. Normal peak shape is seen when the interfering peptide is not present (Figure 1D).
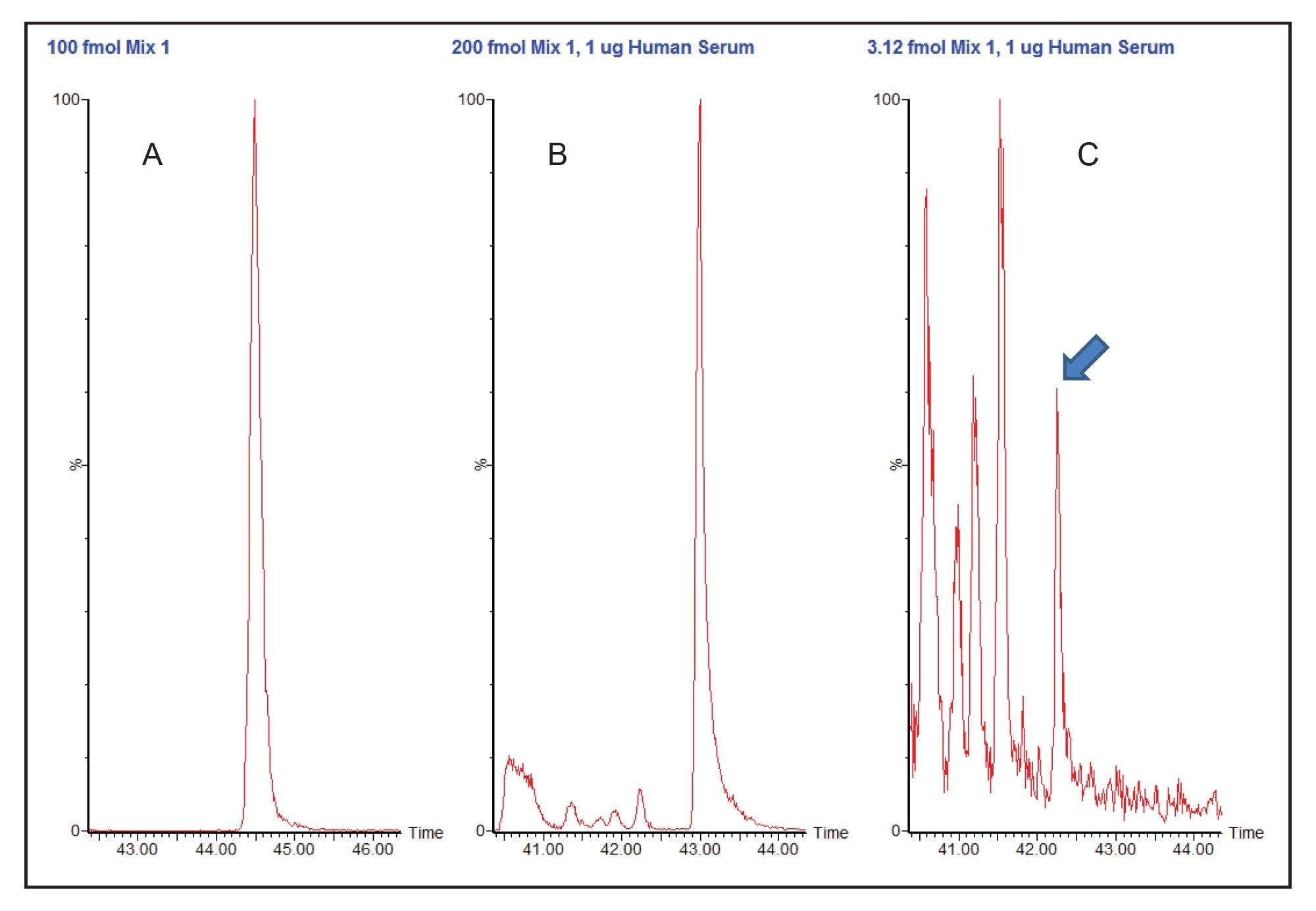
In the case of the peptide DYYFALAHTVR from rabbit phosphorylase b, several additional peaks produced transitions that appeared at retention time similar to that of the analyte peptide. At high concentration (200 fmol, Figure 2B), these additional peaks were small and easily discriminated from the peak of interest. Nearer the limit of detection (3.12 fmol, Figure 2C) the analyte peak detection (arrow) was no longer the largest peak, and it would be more challenging to ensure that the correct peak is chosen for integration and quantification.
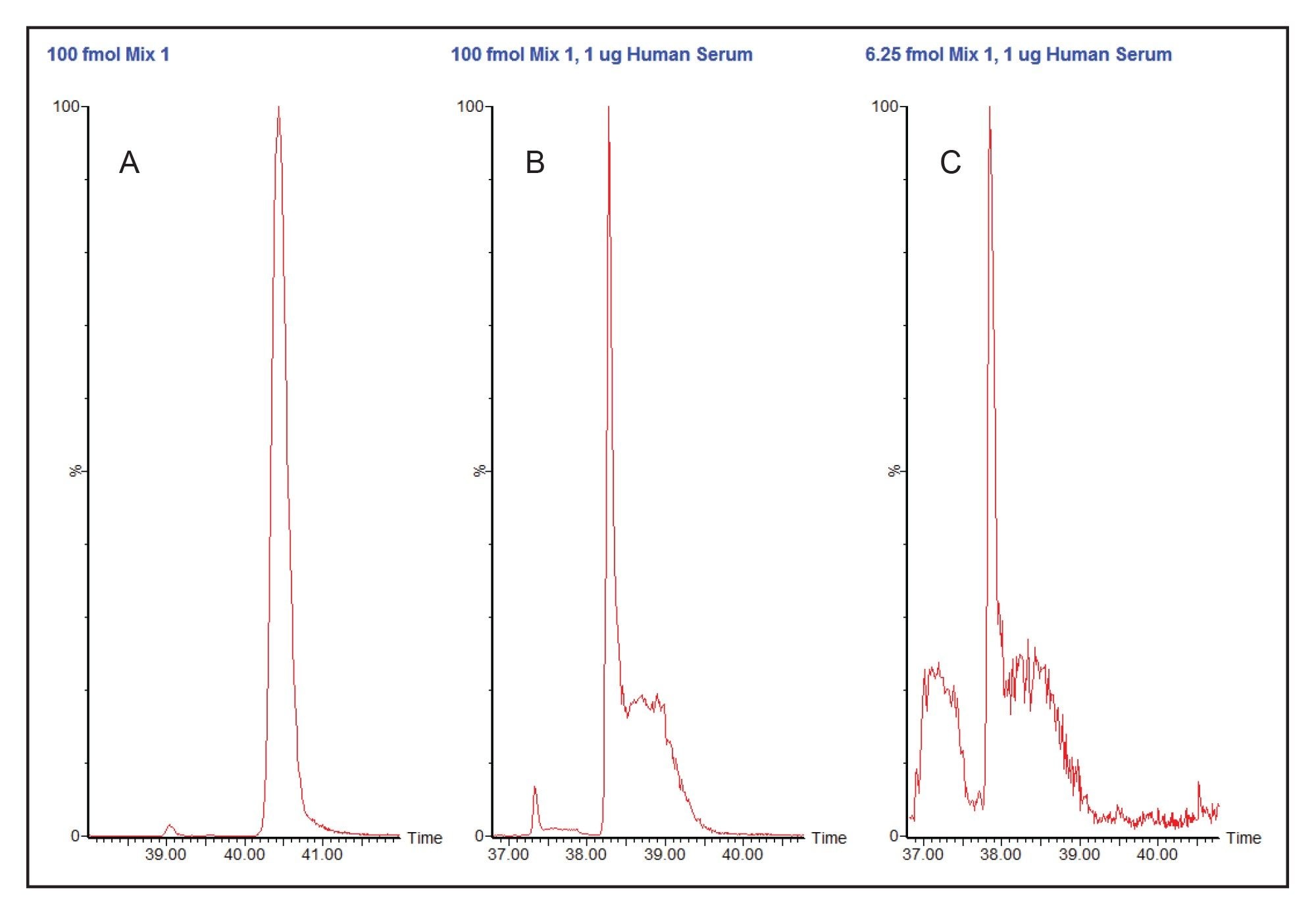
Figures 3B and 3C show what appears to be a coelution of a sharp and a broad peak representing two peptides. Since both of these peaks show the same ratio of broad peak to sharp peak at very different concentrations (100 fmol vs. 6.25 fmol), this is actually a serious perturbation of the chromatography of the analyte by another coeluting but undetected species, very likely a high concentration peptide derived from human serum albumin. The distorted peak would be difficult to integrate accurately.
Conclusion
None of the peptides exemplified here are satisfactory for quantification by tandem quadrupole MRM analysis in the human serum digest matrix. Situations of these types are not uncommon, and care must be taken in method development that peptides selected for quantification are free of interferences and chromatographic perturbations, which would jeopardize the quality of the results. As illustrated in another technical brief,1 when well-behaved peptides are chosen, excellent quantitative results can be obtained.
References
- Dorschel, C.; Favorable Conditions for Quantification of Peptides in Complex Samples by Multipl Reaction Monitoring (MRM) with a Tandem Quadrupole Mass Spectrometer; Waters Application note, 720003883, 2011.
720003884, February 2011