Practical Approaches to Peptide Isolation
Introduction
Peptides play a unique role in the development of new drug candidates and fill a specialized niche between traditional small molecule therapies and larger protein remedies. With higher efficacy, safety, and tolerability in humans as compared to small molecules, and with lower production complexity and cost than protein-based biopharmaceuticals, peptides are increasingly attractive as potential drug candidates targeting many medical conditions. Synthetic peptides may be used to study the structure-activity relationships between cellular substrates or be effective as drugs themselves. Whether peptides are made step-wise using solid-phase peptide synthesis, solution phase synthesis, or isolated from naturally-occurring sources, almost all crude peptide mixtures are complex and require purification. Even though the synthesis reactions are carefully controlled, impurities are inevitably formed and may include deletions, truncations, or chemically-modified sequences including cleavage adducts or other by-products formed during processing.
Technological advances in analytical methodology made in recent years have also influenced the prevalence of peptides as diagnostics and emerging therapeutics. Analytical methods run on state-of-the-art instrumentation show improved sensitivity and higher resolution in a shorter amount of time, both of which lead to critical time savings during production.
Synthetic peptides offer unique challenges in the development of an isolation method. For future studies, purity and target peptide yield are critical factors which influence experimental results. As mentioned above, the number of impurities resulting from the synthesis, cleavage, and deprotection of a synthetic peptide may be quite large. The isolation of the peptide product can be improved by applying method development principles that are often associated with analytical HPLC. The separation mechanisms used to create a purification method at the larger preparative scale are the same as those principles that are utilized at the smaller scale. Column selection, choice of mobile-phase modifier, the use of temperature, and gradient optimization are choices that must be examined in the development of any separation method. Systematic method development for peptide isolations can improve the purity and yield of product significantly. While traditional peptide isolation is usually performed using UV-directed chromatography, using mass-directed isolation along with prudent method development, makes the purification process easier with improved discrimination between the target peptide and the contaminants formed during synthesis and cleavage.
Peptide Purification Process Requirements
The goal of any isolation strategy is to purify as much sample in as little time as possible with the purity required to perform subsequent experiments. For the typical laboratory that synthesizes many different peptides, it is useful to establish a standard purification protocol that can be used for most samples, while permitting simple optimization when necessary. Changes in mobile-phase composition, modifier, or temperature on columns with consistent run-to-run performance eliminate the need to have multiple column sets for isolation. These economical options for improving yield and purity are straightforward and easy to implement. The optimal peptide purification process also utilizes robust instrumentation capable of executing isolations at a range of analytical and preparative scales. Merely changing the column to the appropriately-sized column diameter for the isolation on a versatile instrument with wide flow rate range makes purification efficient. All of these factors, considered together, can reduce the overall costs associated with peptide purification.
Peptide Isolation Workflow
A standard peptide isolation protocol (Figure 1) begins with defining how much pure peptide is required to perform ensuing experiments. Crude peptide analysis provides a reasonable estimate of sample purity, and combined with the defined amount of pure peptide required for later studies, the estimated mass load can be calculated. The mass load defines the prep column size and, in turn, the flow rate required on the selected LC instrument.
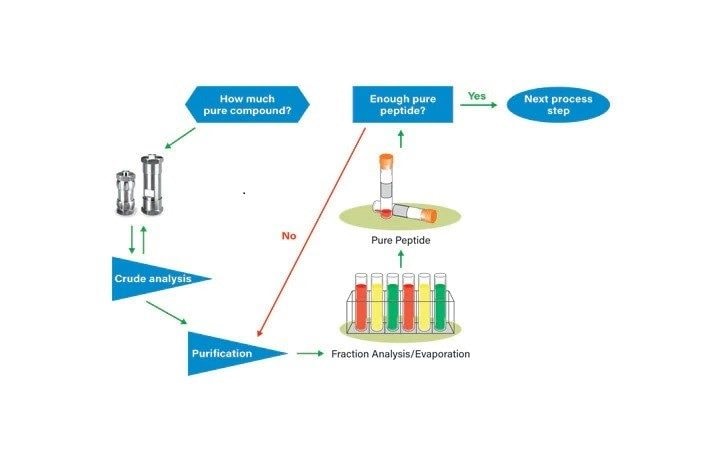
Geometric scaling to a large column of the same chemistry for isolation follows crude peptide analysis. The scaling equations in Figure 2 show the relationships between column diameter, column length, mass loading, and flow rate for moving a separation from one column size to another. Expressing gradient slope as % change per column volume, instead of % change per unit time, assists in ensuring that resolution is preserved by delivering the same number of column volumes for each gradient method step from initial to final conditions. Preparative methods must include an initial hold at starting conditions to mimic the system delay, also expressed in number of column volumes, which is observed on the small scale separation. Once the preparative chromatography is completed, the collected fractions are evaluated analytically. Pure peptide is subsequently processed as defined by user guidelines.

The simplest liquid chromatography systems configured with a pump, injector, UV detector, and a fraction collector are perfectly suitable for peptide isolation (Figure 3). Mass detection, although not required for peptide isolation, identifies the peptide target and reduces the number of collected fractions requiring analysis after isolation. Compounds that do not ionize, or ionize poorly, will often be detected with low wavelength UV. Conversely, peptides with very low UV extinction will usually be readily detected with MS.

Purification Process Considerations
Separation Modes
Because the properties of different peptides are so diverse, the chromatographic modes of separation also vary. While reversed-phase chromatography is, by far, the most popular mode for peptide purification, some peptides are more efficiently isolated using an alternative selectivity such as ion-exchange or size-exclusion. These alternative techniques may also be combined with reversed-phase, creating a two-step process for difficult samples.
Reversed-Phase
Because of its reproducibility and broad applicability, reversed-phase chromatography is commonly used for the separation of many types of compounds, including peptides. C18 –bonded silica, one of the most popular types of reversed-phase column packing, is non-polar. Reversed-phase mobile-phases, usually comprised of water with a miscible polar organic solvent, such as acetonitrile or methanol, elute compounds from C18 columns based on their polarity. Therefore, the more hydrophobic in nature a peptide is, the more it will retain on a C18 column.
Although C18 is the most popular reversed-phase packing, other ligands (C4, C8, RP18, Phenyl, and others) can also be bonded to the base particle to provide alternative selectivity for the optimized separation of compounds. While silica was one of the first substrates to be used for reversed-phase packings, silica-based packing materials impose limits on separation speed, resolution, pH, temperature, and column loading capacity. The patented* hybrid particle technology, used in the development of new column substrates, creates particles that can be operated at higher speeds, temperatures, and pH while improving selectivity, resolution, reproducibility, and column lifetime.
*U.S. Patent No. 6,686,035 B2
Ion-Exchange
Ion-exchange chromatography has long been employed for the isolation of proteins and other biologics. Because the amino acids in a peptide can be either positively or negatively charged, ion-exchange chromatography can be used for peptide isolation. Functionalized natural polymeric resins such as agarose, synthetic polymers like poly(methyl methacrylate) (MMA), or polystyrene divinylbenzene are commonly used media for large molecule separations, since these materials can withstand the rigorous cleaning requirements that must occur between process cycles and batches in large scale bioprocessing. Sodium hydroxide solutions utilized in these processes are deleterious to silica-based materials commonly used for small molecule and small scale peptide isolations.
There are four types of ion-exchangers: weak anion, weak cation, strong anion, and strong cation. Cation-exchange is used to retain and separate positively charged ions on a negative surface, while anion-exchange is used to retain and separate negatively charged ions on a positive surface. Strong anion and cation-exchange columns are completely ionized over a broad pH range (2–12), whereas weak ion-exchange columns are charged over a narrow pH range (9.5 to 5.5).
Peptide isolation using cation-exchange is more common than anion-exchange, but the preferred mode really depends on the peptide sequence. For peptide separations performed at a pH lower than 3, the negative charges on the amino acidic side chains carboxyl groups are neutralized and the N-terminal groups are protonated, making the peptides positively charged and attracted to the negative sites on the cation-exchange column (Figure 4).
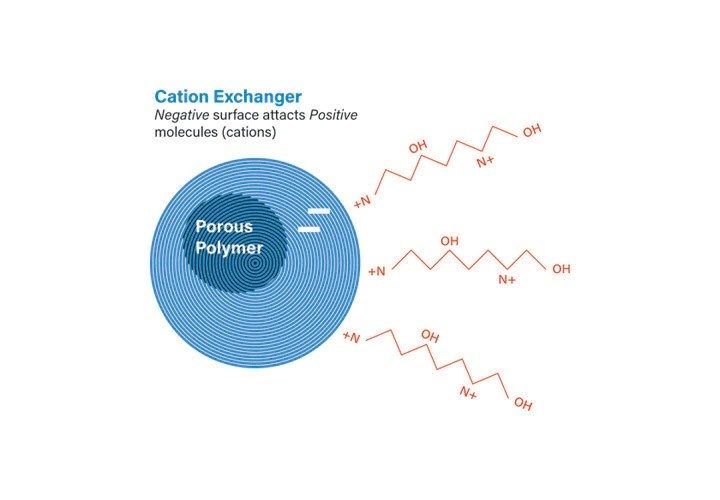
Conversely, for those peptides more suited to separation at between pH 6 and 10, anion-exchange may be used. With anion-exchange at higher pH, the carboxylic groups on the peptide are negatively charged and attracted to the positive sites on the anion-exchange column (Figure 5).

Some general suggestions can be offered for selecting a column and developing a method for peptide isolation using ion-exchange. Either anion or cation-exchange can be used. As a general starting point, acidic peptides are separated on anion-exchange and basic peptides on cation-exchange. This selection may be usefully reversed for some peptides, particularly larger ones, but the pH must be adjusted so that the net charge is the opposite of the packing material.
For either polarity, strong exchangers are preferred for peptide chromatography. The charge on a peptide is a function of the sequence and the microenvironment around the charged side chains. Small adjustments in mobile-phase pH can be used to adjust separation selectivity based on the specific peptide sequence. These small adjustments to selectivity do not affect the binding capacity of the strong ion-exchanger.
Selection of the mobile-phase and operating conditions is influenced by whether the ion-exchange separation is planned to be the first step of a two-step purification or if it is to be the single isolation step that produces experimental material. If the material collected from ion-exchange is to be further purified with reversed-phase, the buffers and elution conditions can follow the usual practices for protein ion-exchange; for example, using phosphate or Tris buffers and eluting with a gradient of sodium chloride. The salts will be removed from the product in the subsequent step.
If ion-exchange is chosen to produce usable peptide in a single step, it is prudent to choose volatile buffers that can be removed by lyophilization. Ammonium formate is a good first option, since it has two buffering regions corresponding to the pK’s of the ammonium and the formate. It can be used from pH 3 to pH 4 for cation-exchange chromatography of basic peptides or from pH 9.25 to 10.25 for anion-exchange chromatography of acidic peptides. Elution can be effected with an ionic strength gradient of increasing ammonium formate concentration at constant pH. Alternatively, elution with a pH gradient reduces time-consuming lyophilization from high ionic strength buffers. Again, ammonium formate buffers provide a useful starting point. Elution proceeds from high to low pH for acidic peptides on anion-exchangers and from low to high pH for basic peptides on cation-exchangers.
Size-Exclusion
The chromatographic separation of molecules based on size, called size-exclusion chromatography (SEC) today, dates to the 1950s. The separation of molecules based on size, rather than on their chemical properties such as charge or polarity, has also historically been known as gel filtration or gel permeation chromatography (GPC). Stationary phase particles with a defined pore size distribution exclude sample molecules that are larger than the cavities in the column bed, causing them to elute very quickly, while small molecules enter the pores and, subsequently, navigate a more tortuous and slower path to elution. Therefore, large molecules elute first, while smaller molecules elute in decreasing order of their size in solution (Figure 6). SEC is a low resolution technique which utilizes isocratic methods and requires no equilibration between runs. Column loading is dependent upon the sample volume and concentration, which both affect resolution.
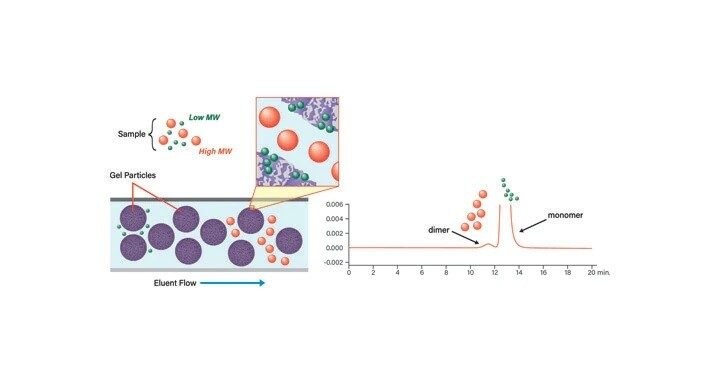
SEC is sometimes used as an initial cleanup step to remove truncated sequences, peptides with incomplete deprotections, and other impurities. As a synthetic peptide grows in length, the number of these contaminants may be significant. Any simple initial purification step prior to isolation by reversed-phase reduces chromatographic complexity. SEC can also be used for buffer exchange, that is, changing the buffer from which the peptide is currently dissolved to another buffer or solution that is more suitable for the next experiment or purification step.
Summary
Peptides can be analyzed and isolated using many different chromatographic techniques including reversed-phase, ion-exchange, and size-exclusion. The methodology employed at the semi-prep scale (a few milligrams to a few hundred milligrams for isolation) is typically reversed-phase. The remainder of this discussion on peptide isolation will be devoted to the factors and techniques that can be used to optimize peptide analysis and purification using reversed-phase chromatography.
In this Primer
Practical Approaches to Peptide Isolation
Method Development Considerations