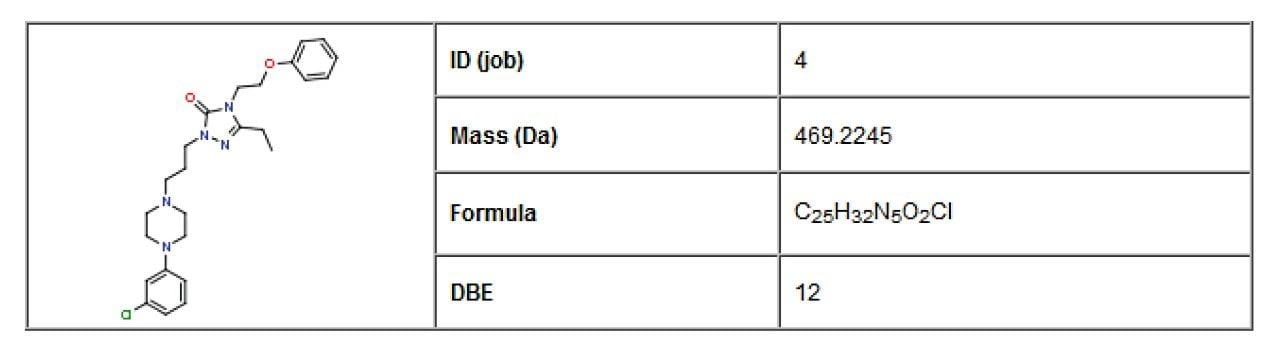
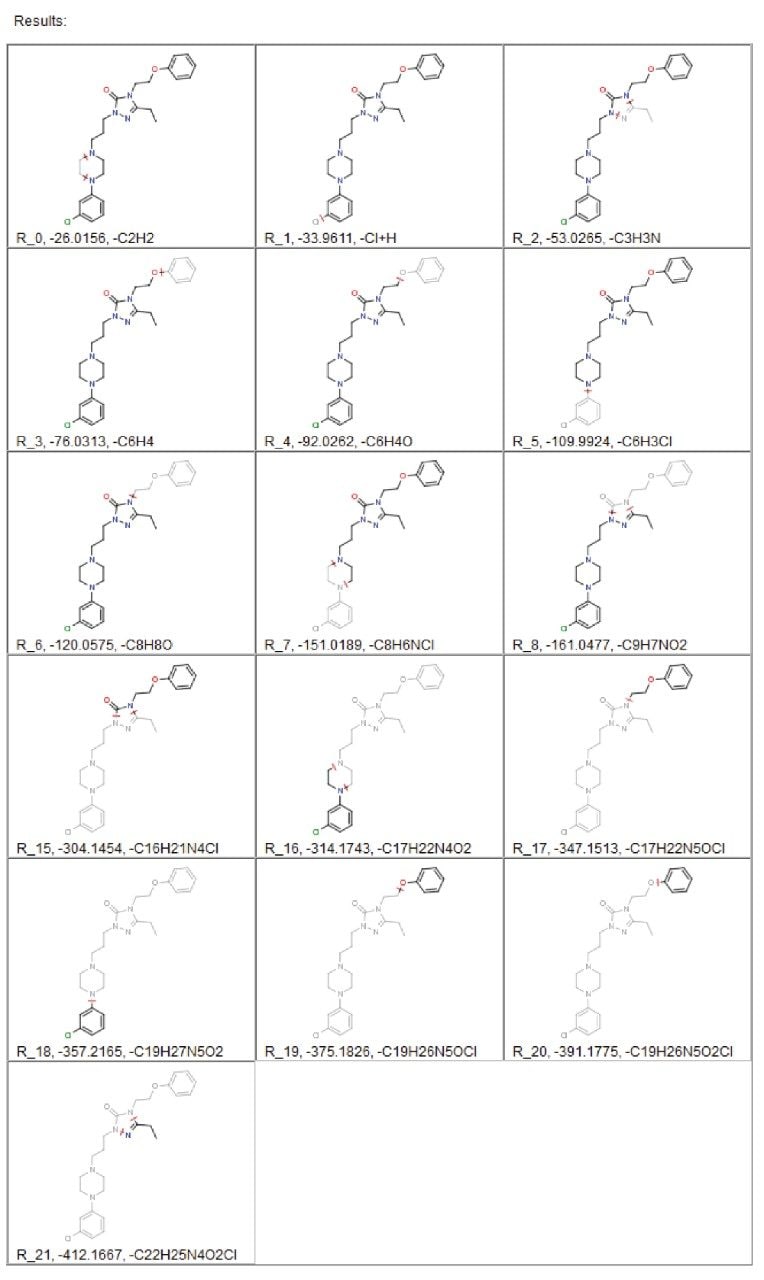
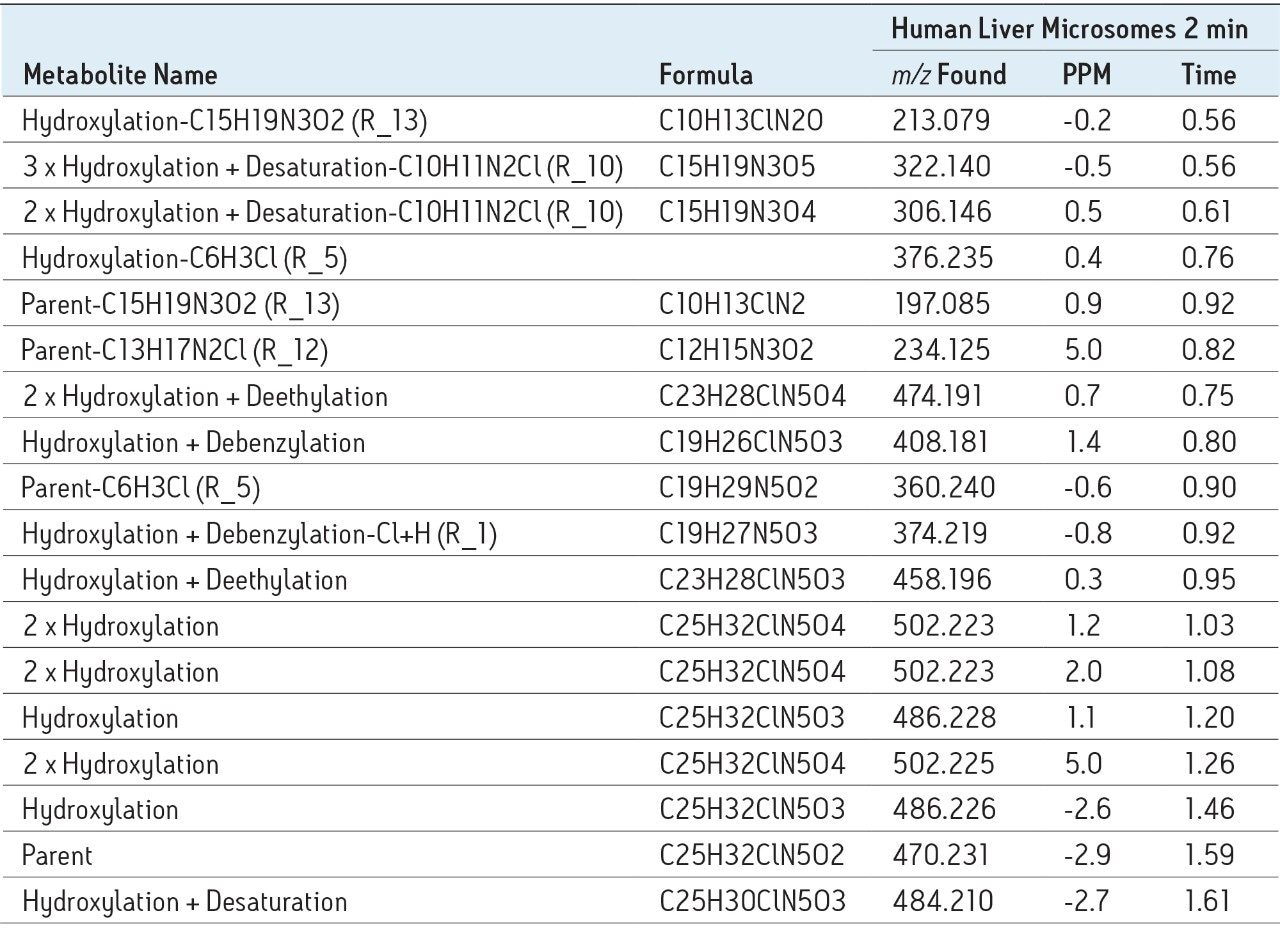
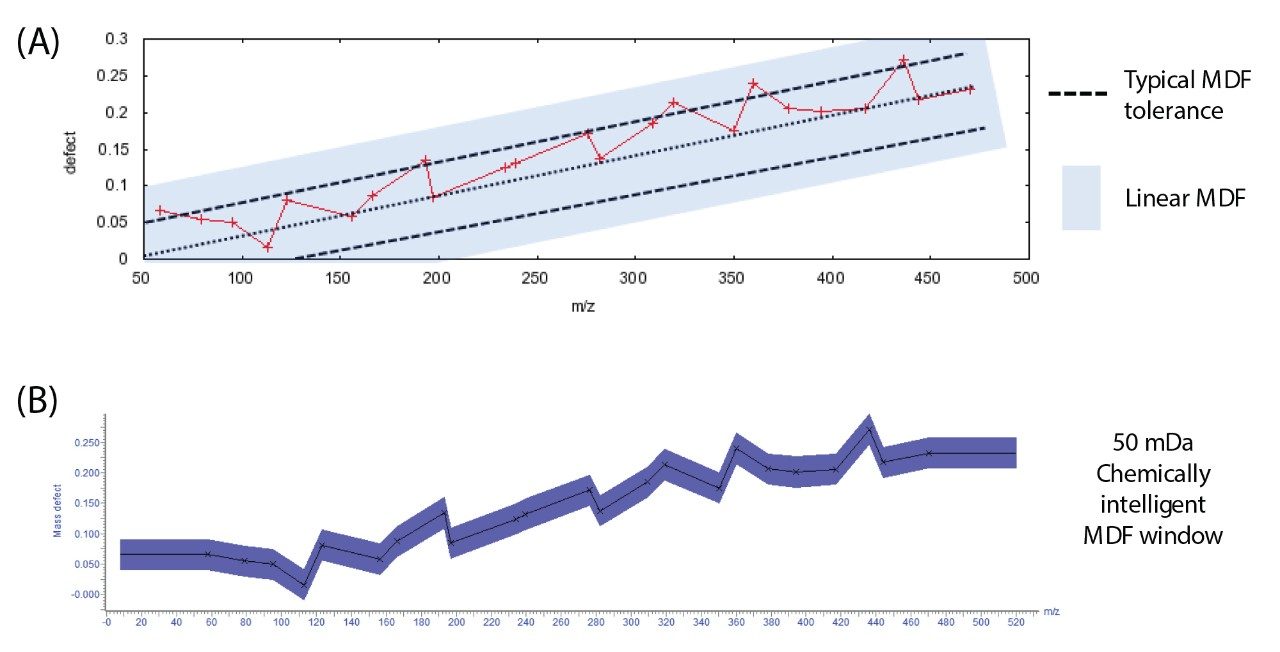
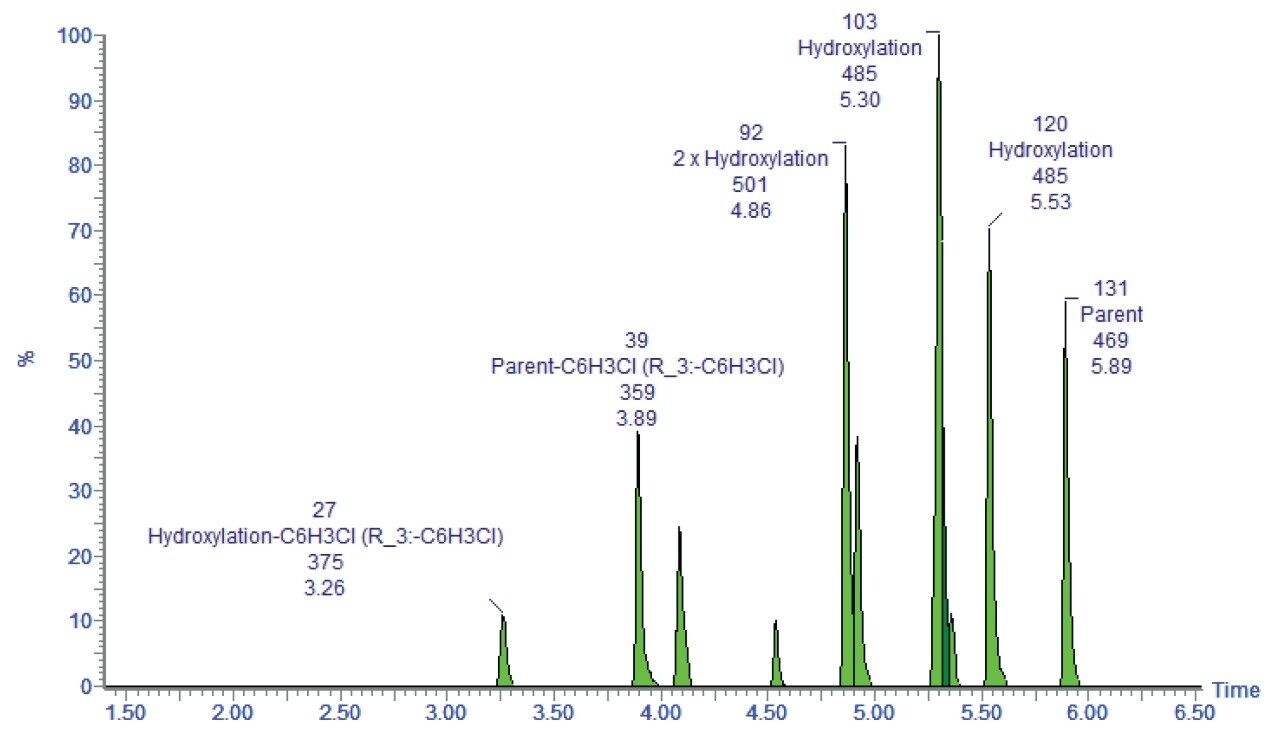
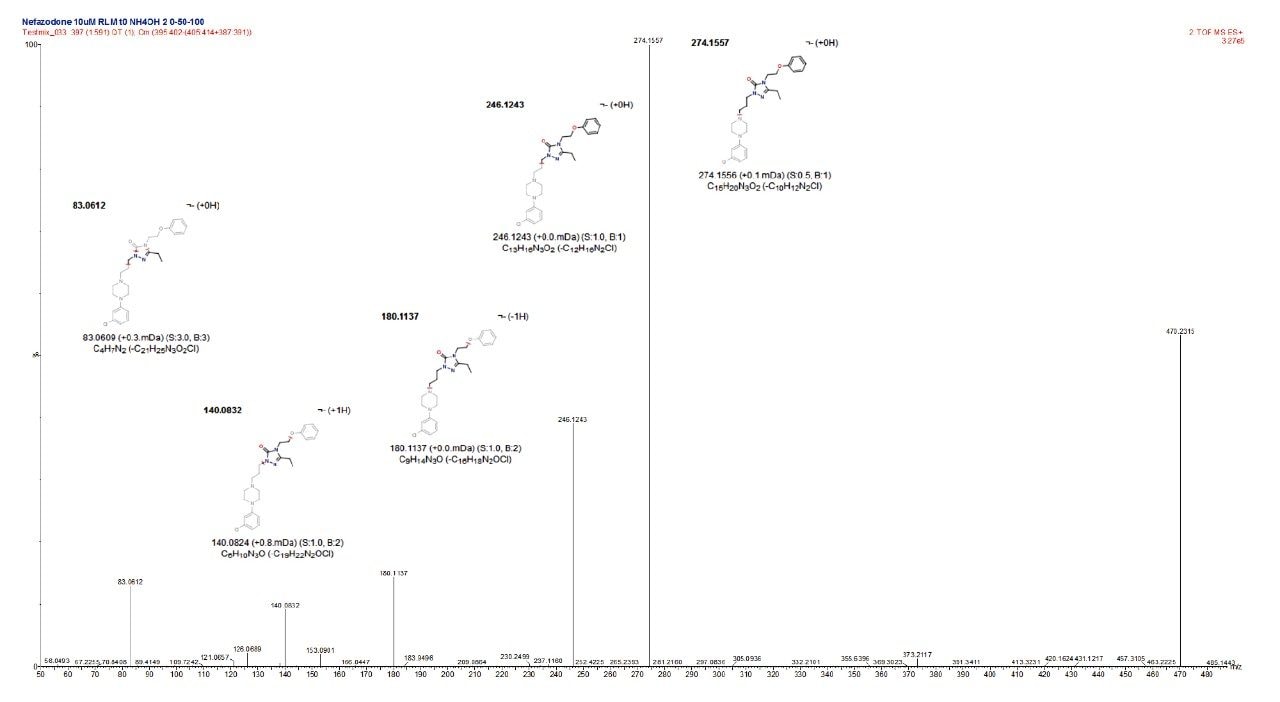
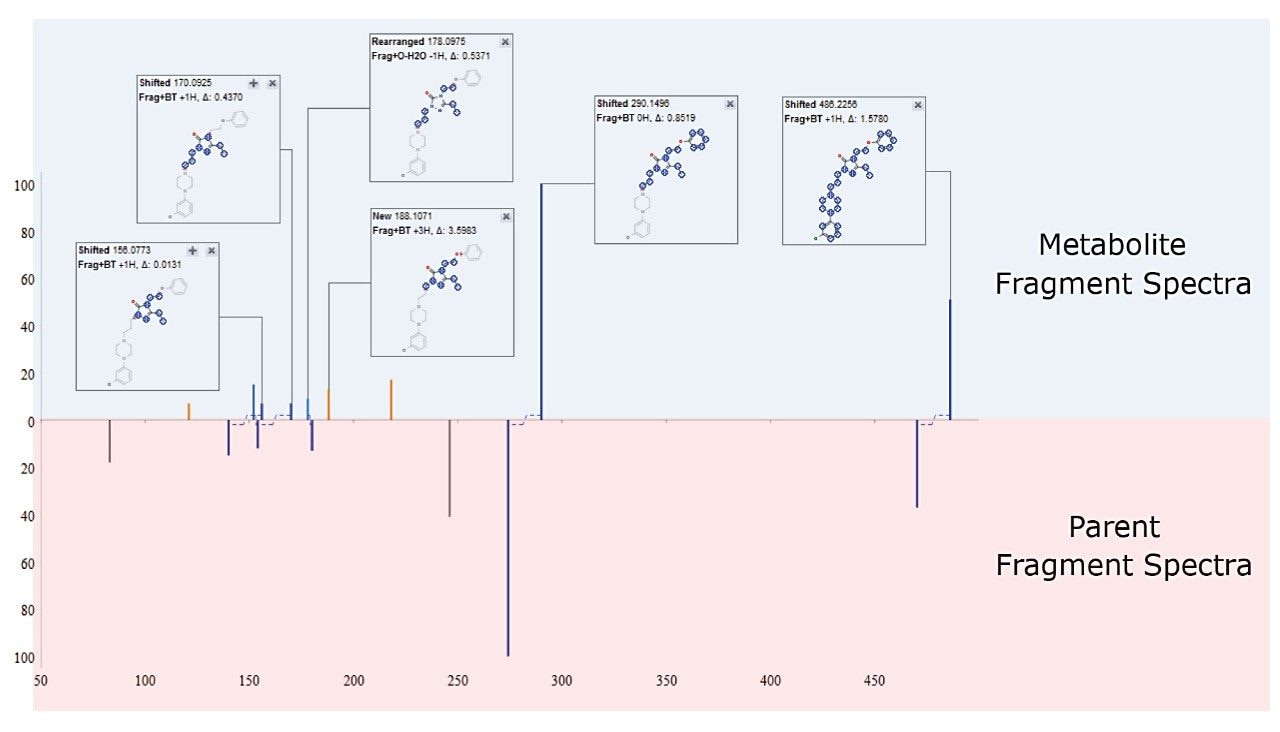
A large number of routes of metabolism have been characterized by the scientific community and this has yielded a diverse knowledge base of commonly occurring metabolic transformations. The first line of metabolite identification is to proactively search for these known pathways. This knowledge combined with the exact mass measurement instrumentation has facilitated the positive identification of these metabolites in a routine fashion. This is a powerful tool in the arsenal of the drug metabolism chemist and many metabolites have been positively identified in this manner. However, for new drug entities, drug metabolism is driven by reactions that occur between the drug structures and the enzymes responsible for drug metabolism. These very specific interactions often spawn metabolites that are cleavage products of the starting structure and therefore cannot be fully understood by mining a database containing metabolites derived from a set of known compounds. If these unique biotransformations are not taken into consideration, many novel metabolites may not be positively identified. The combination of structural knowledge and metabolite identification tools is typically not used to direct metabolism identification studies up front.
In this application note, we will demonstrate the use of chemical intelligence to broaden our approach to metabolite identification. This results in an analysis that gives more confident answers in a more efficient workflow, allowing the drug metabolism scientist to output results more quickly and effectively to drive key program decisions.