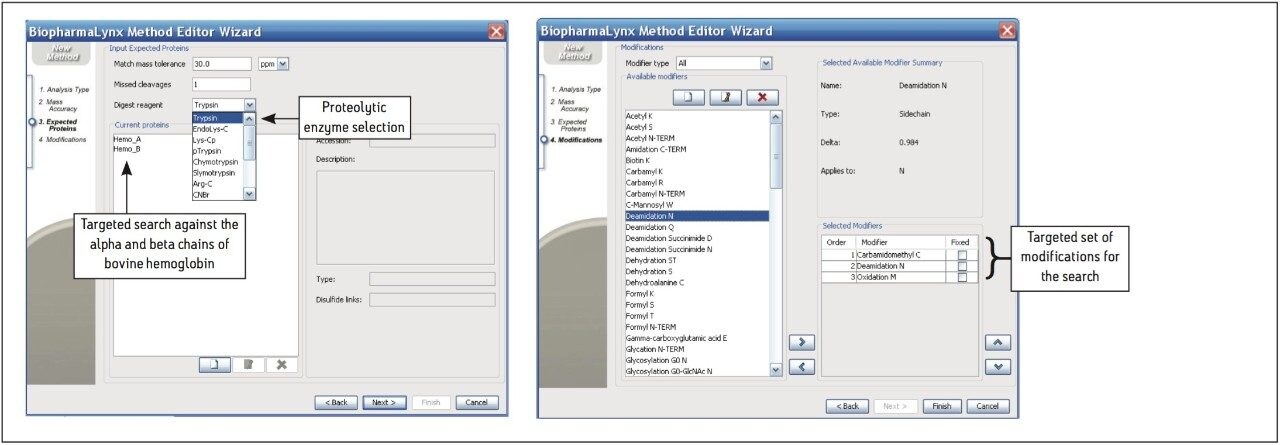
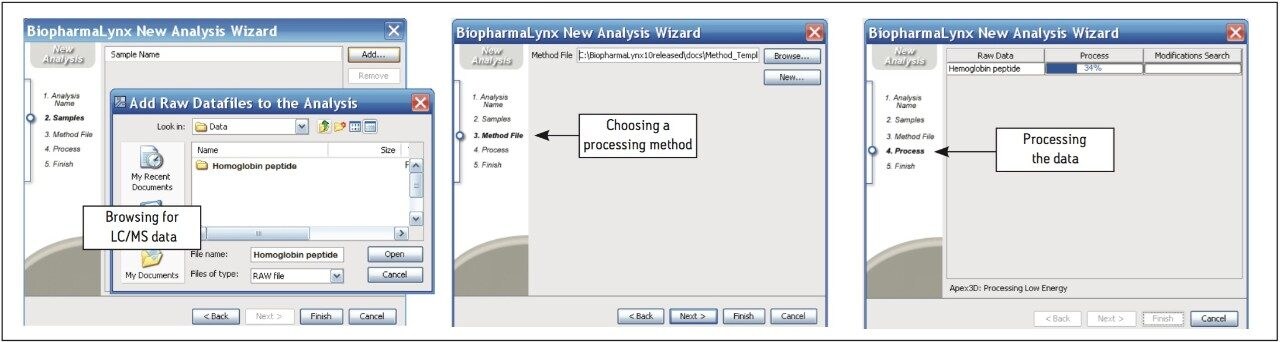
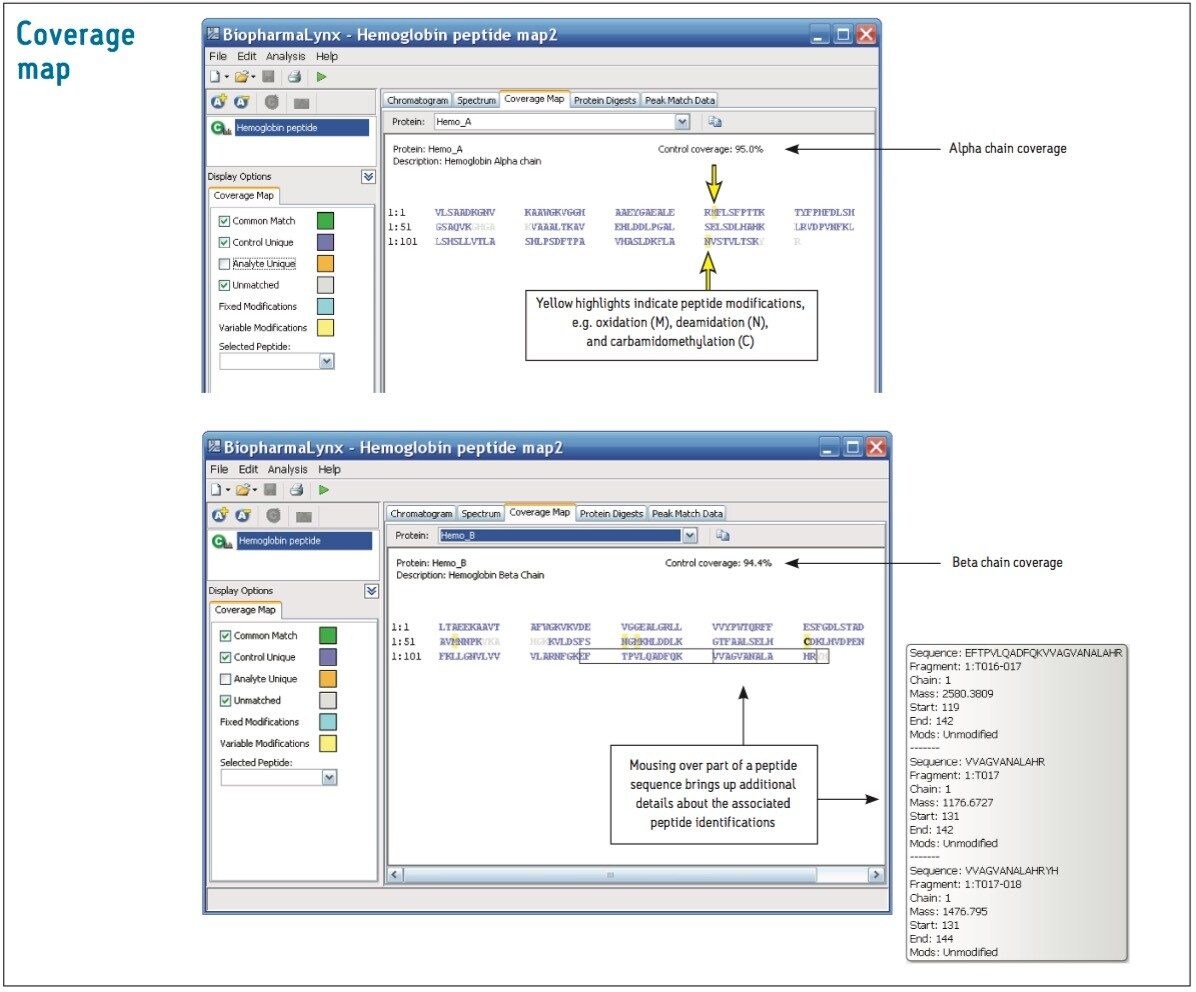
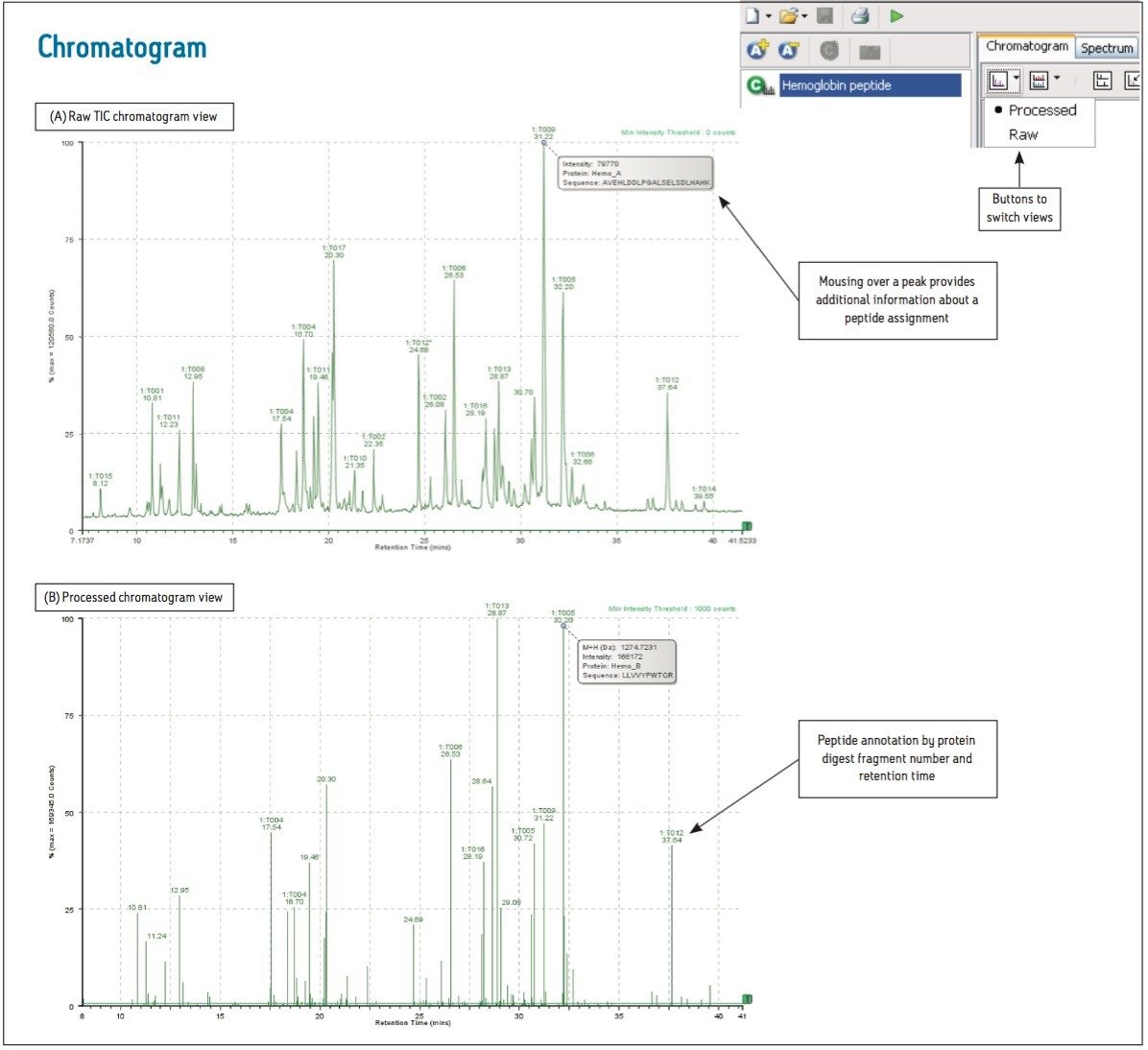
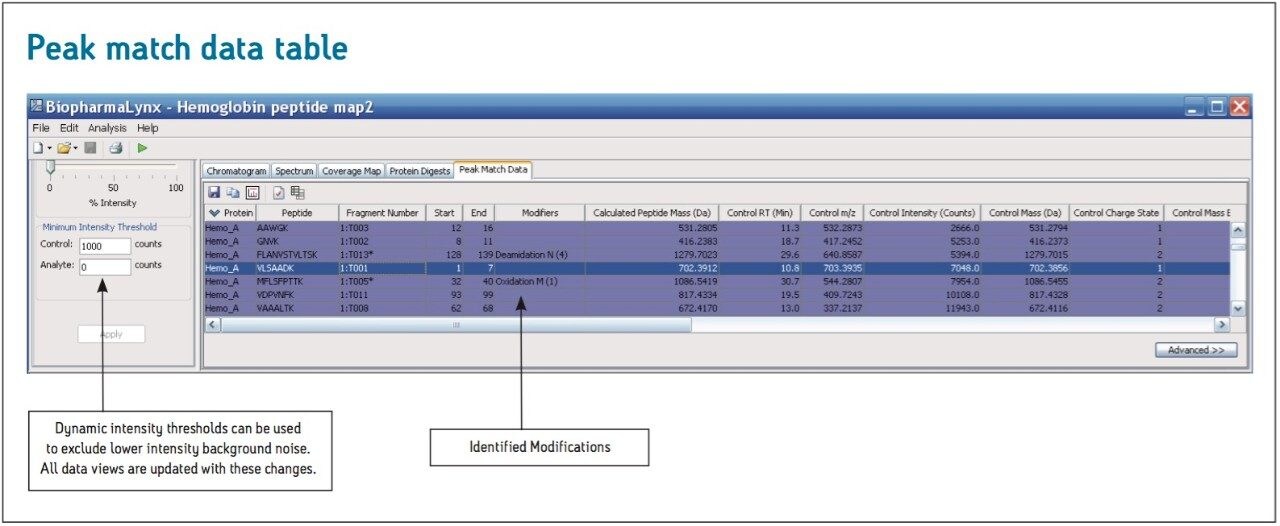
The BiopharmaLynx Application Manager is a powerful bioinformatics tool that automates data processing and peptide annotation for LC-TOF/MS peptide mapping data sets. Data browsing with graphical and tabular tools quickly provides the information needed to make decisions about protein quality or effectiveness of an analytical method or process. Obtaining better information faster can decrease development costs and streamline development timelines.
Waters provides a total system solution for peptide map analysis that includes ACQUITY UPLC-based separations to enable faster more resolving peptide maps, multiple UPLC chemistries to exploit map selectivity differences, and sensitive TOF mass spectrometers for accurate mass and modified peptide identifications. BiopharmaLynx complements Waters peptide mapping solutions, allowing for integrated peptide mapping data analysis with greater speed and confidence. Automated processing and annotation of peptide maps can be achieved in minutes, removing the most tedious and productivity-limiting element of a peptide mapping analysis.
In conclusion, BiopharmaLynx decreases the expense and time-tomarket of protein therapeutics by reducing data analysis time for peptide mapping studies. This software offers fast data processing and an easy-to-use interface that saves time and allows scientists to engage in other high-value tasks.