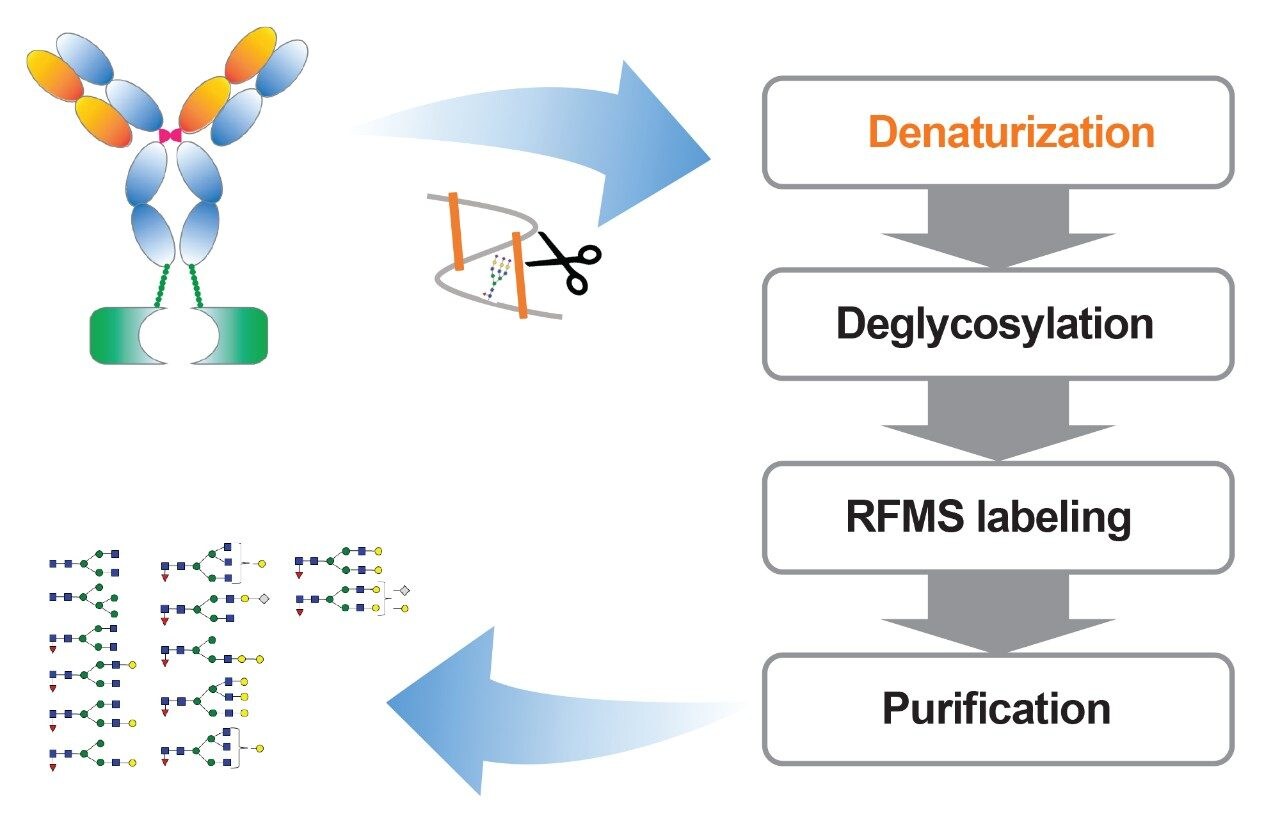
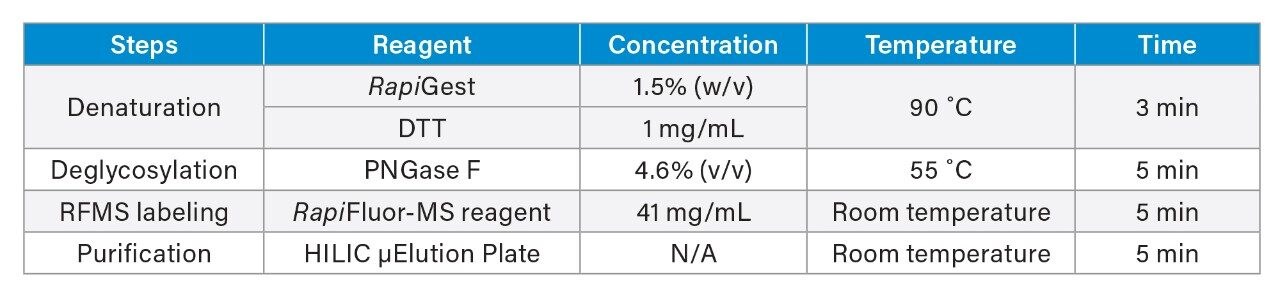
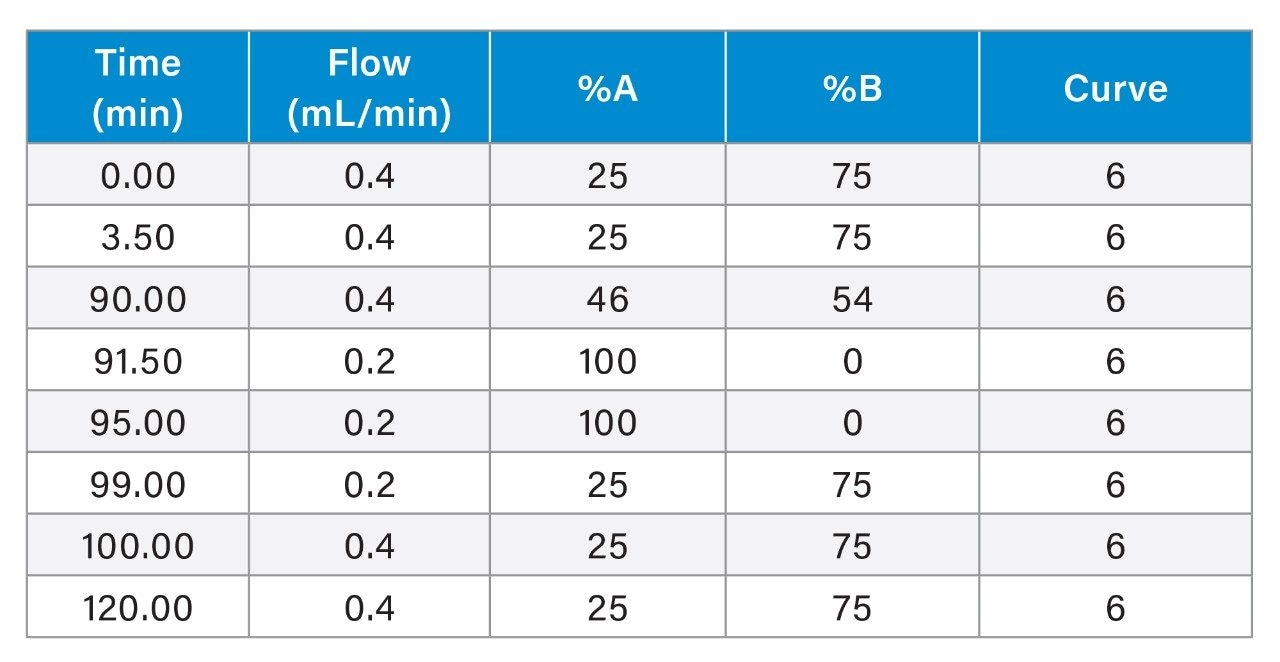
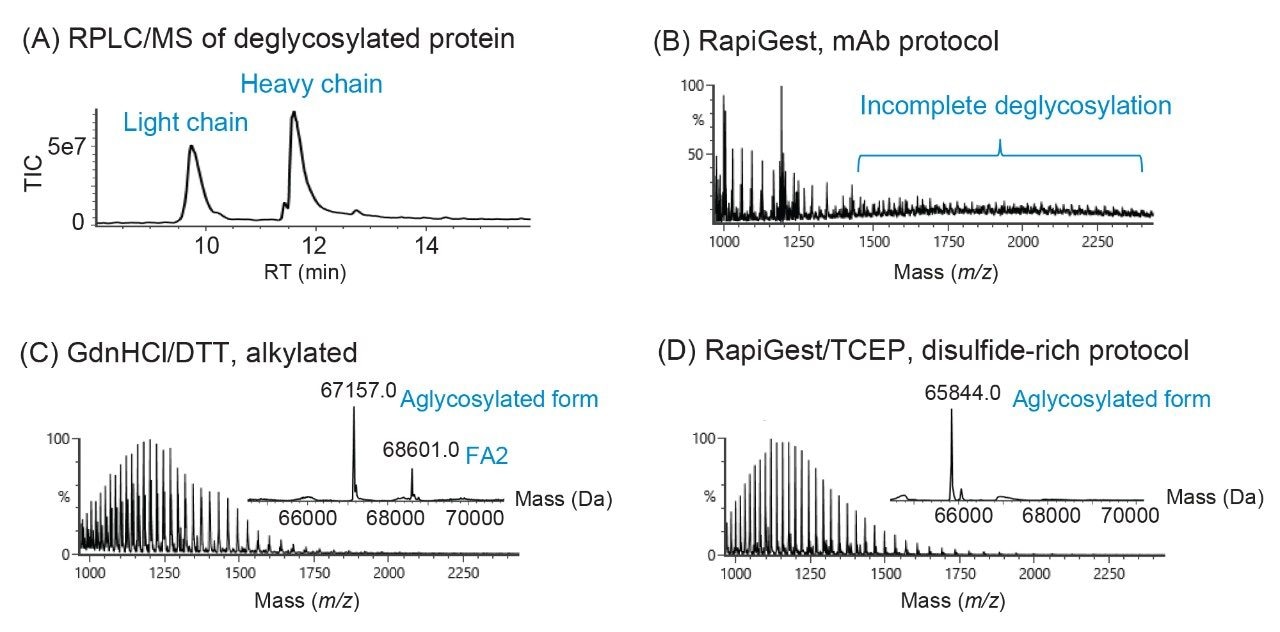
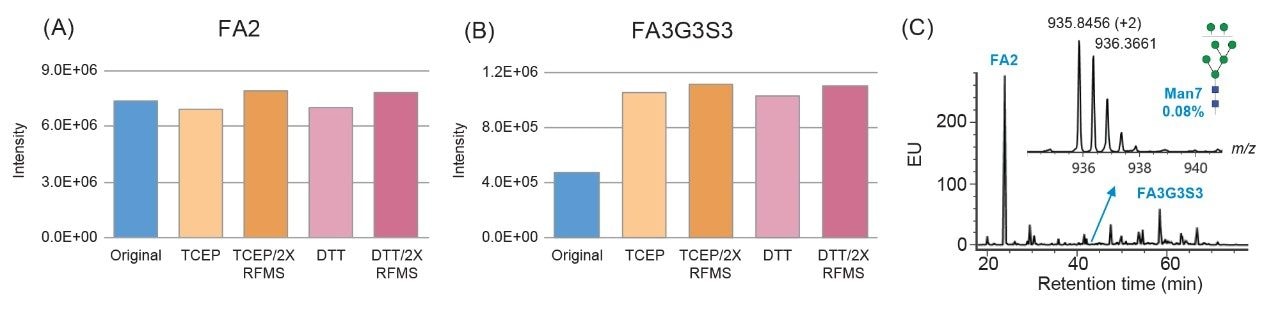
To expand the usability of the sample preparation method in upstream and downstream activities, the effect of a more commonly used reducing agent, DTT, was evaluated on the labeling efficiency and yield of glycans. Although the thiol group on DTT can react with the RFMS reagent and affect glycan labeling, a 6.5 mM final concentration of DTT was found to have a similar impact on labeling and glycan releasing compared to that of TCEP (Figure 3A and 3B). Since DTT does not need neutralization prior to use and is typically more cost-effective, it would be more QC-friendly to be used as the reducing agent for sample preparation in a routine analysis environment. As mentioned previously, the incorporation of a reducing agent in the denaturing step allowed for increased accessibility of the PNGaseF enzyme to solvent restricted glycan sites. This resulted in the increased yield of N-linked glycans and detection of low abundance glycans in both FLR and MS as noted by the high signal-to-noise ratio obtained for Man7, a glycan at 0.08% relative abundance (Figure 3C). Together, these results demonstrated the improved accuracy and efficiency of the sample preparation method for N-glycan profiling of disulfide-rich and heavily glycosylated fusion proteins.
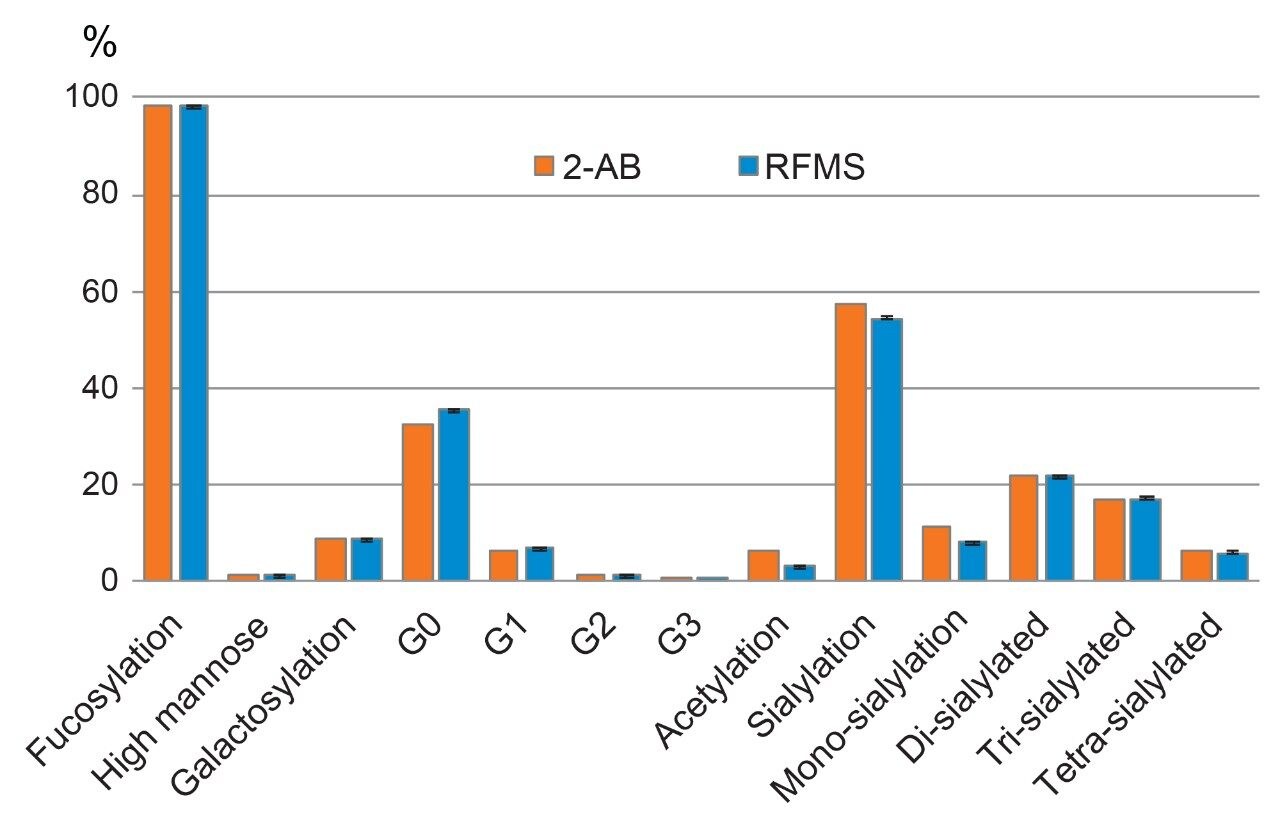
Evaluation and adoption of new technologies are recommended by the International Committee of Harmonization (ICH) to ensure product quality and safety, which emphasizes the importance of comparability studies in facilitating the transfer from legacy methods. In the biopharmaceutical industry, conventional labeling reagents such as 2-aminobenzoamide (2-AB) and 2-aminoanthranilic acid (2-AA) have been widely used in glycan derivatization for FLR detection.7 While effective in obtaining sensitive FLR response, these methods require long enzymatic deglycosylation and FLR labeling time and use complicated sample preparation protocols. Scientists in the biopharmaceutical industry have been looking for alternative methods that are efficient, easy to follow, and sensitive. To evaluate the comparability of N-glycan analysis results using different sample preparation methods, the glycans prepared using the optimized RFMS-based method were separated and quantified based on the relative peak area in FLR chromatogram, and then grouped based on the glycan structural attributes. As shown in Figure 4, a 98.3% fucosylation, 54.3% sialylation, and 1.2% high mannose type glycans were obtained using the optimized RFMS sample preparation method. These results were overall comparable with the data previously obtained from the conventional labeling method that employed GdnHCl/DTT denaturation and 2-AB labeling, demonstrating the feasibility of migration from legacy 2-AB labeling method to the optimized RFMS method. In addition, a higher level of FA2 (or G0F, in the group of G0) was observed in the RFMS derivatization method, which was consistent with the observation in Figure 2C where FA2 was not fully released in the GdnHCl denaturation method. Collectively, these results demonstrated that this sample preparation can be easily adapted to the optimized RFMS-based method for N-glycan analysis of disulfide-rich fusion proteins with improved deglycosylation efficiency and overall accuracy for glycan profiling.