Methods
One hundred microliters of whole blood was precipitated in two stages. First, 200 µL of isopropanol (IPA) containing 50 ng/mL deuterated internal standard was added and vortex-mixed for 5-10 seconds to fully mix the sample. Next, 800 µL of ACN containing 0.1% formic acid (FA) was added immediately, and the samples were once again vortexed. The samples were then centrifuged at 21K rcf, for 10 min, and the supernatant was loaded directly onto Waters Ostro Pass-Through Sample Preparation plates. Samples were eluted with 2 x 400 µL aliquots of 60:20:20 ACN:IPA:water. Twenty microliters were analysed using an ACQUITY UPLC I-Class PLUS System (FTN) in combination with a Xevo TQ-S micro Tandem Quadrupole MS.
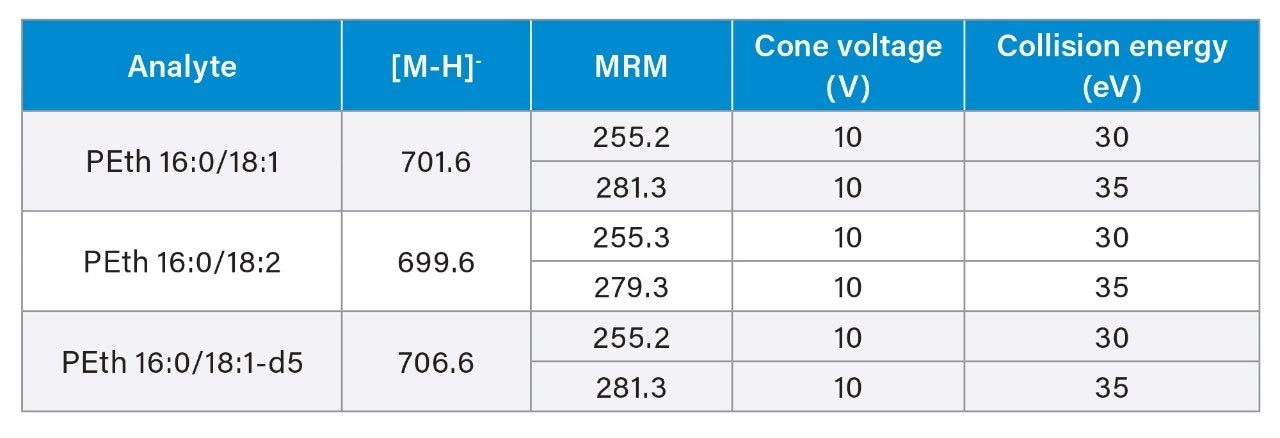
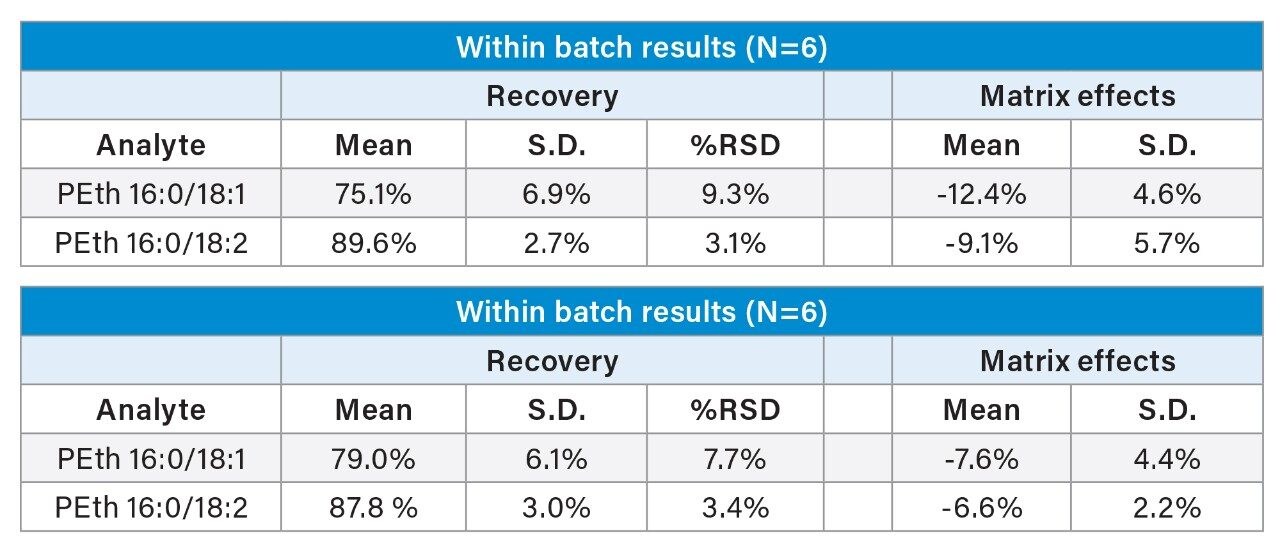
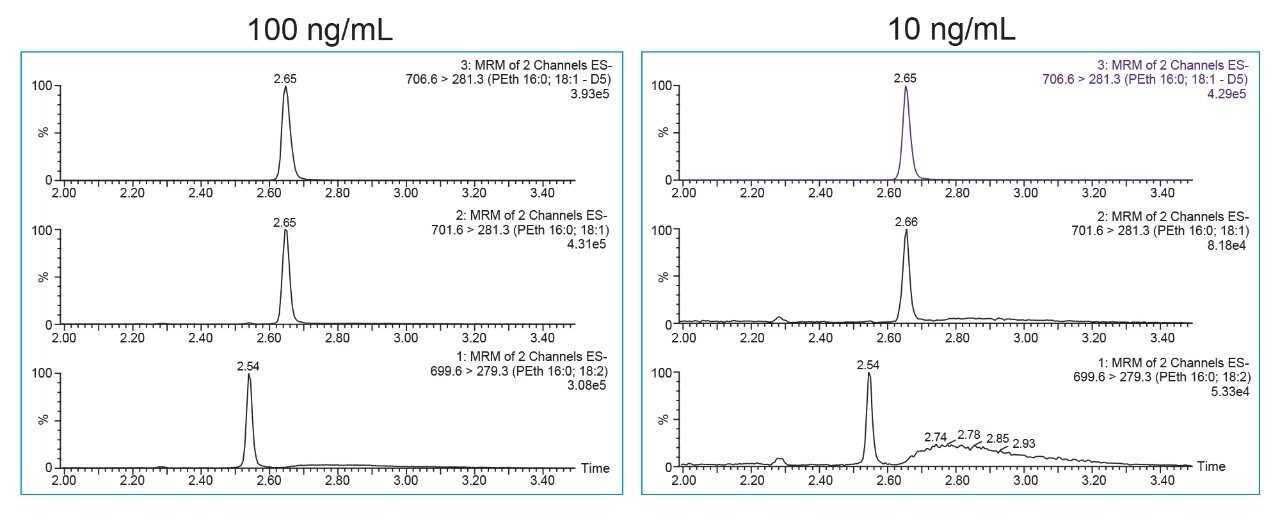
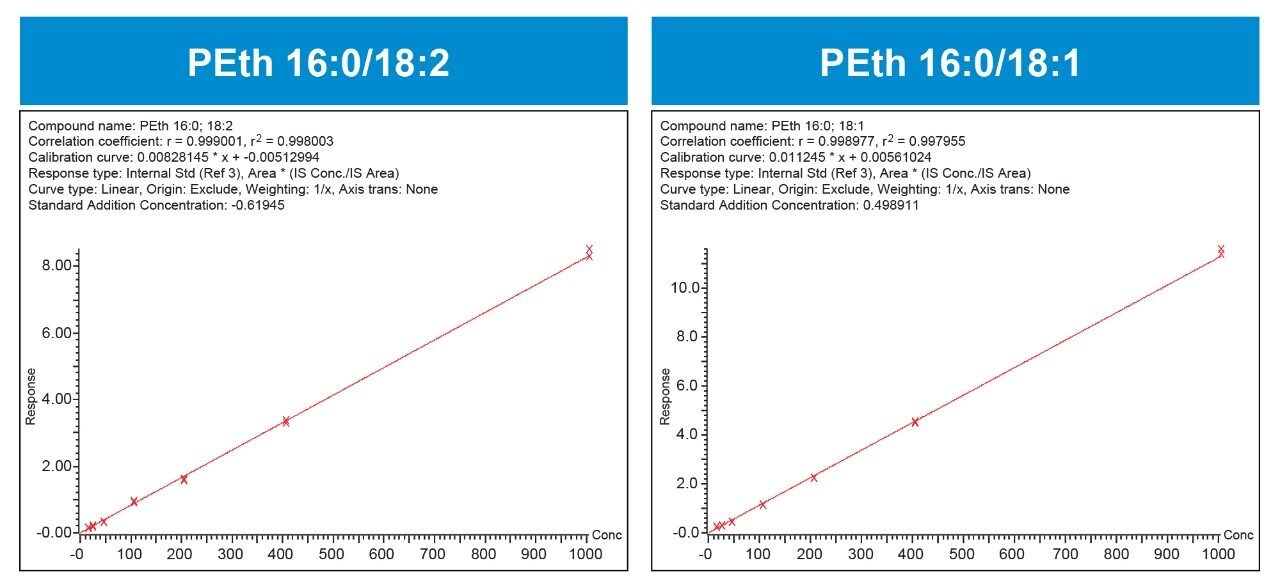
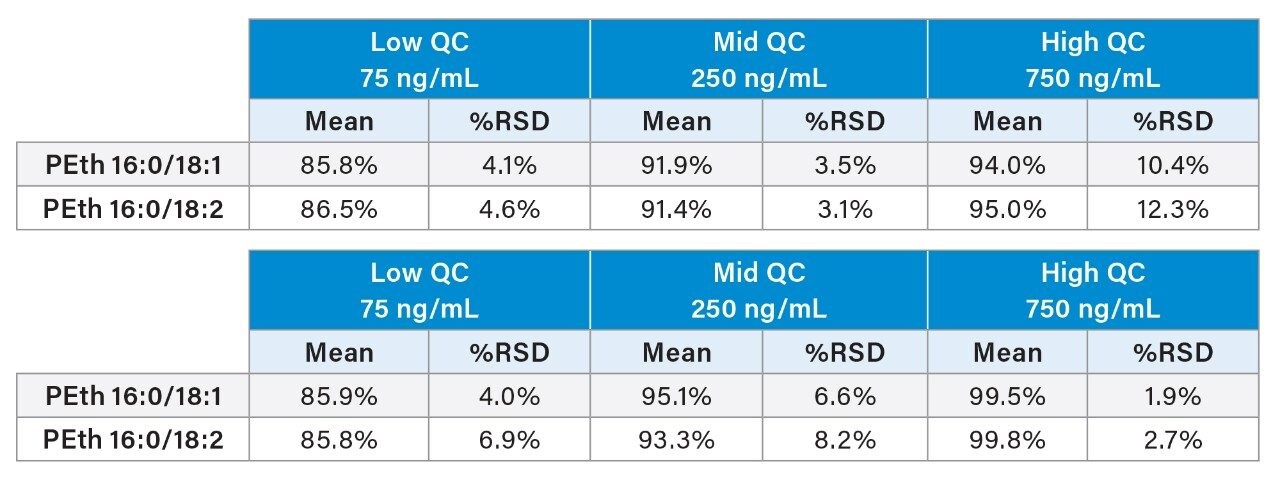
Analytes were chromatographically separated on a 1.7 µm Waters BEH C8 Column (2.1 x 50 mm) at 40 °C. Mobile phase A (MPA) was 5 mM ammonium formate with 0.1% formic acid; mobile phase B consisted of 50:50 ACN:IPA. The flow rate was 0.5 mL/min. The solvent ramp started at 50:50 MPA:MPB and increased to 100% MPB over three minutes. Two rapid 30 second ramps from 50:50 MPA:MPB to 100% MPB were added after the analytical run to minimize carryover. Samples were analyzed in negative ESI mode. Two transitions were monitored for each compound. MS parameters are listed in Table 1. Calibration curves ranged from 10–1000 ng/mL (0.014–1.4 µM). The method was validated for extraction recovery, matrix effects, linearity, accuracy, precision, analytical sensitivity, carryover, dilution integrity, and extracted sample stability.