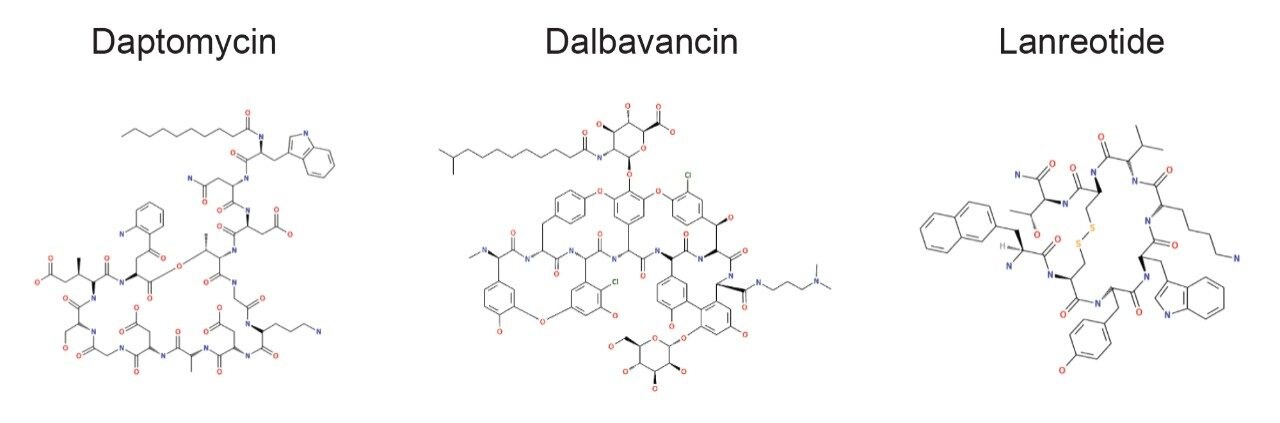
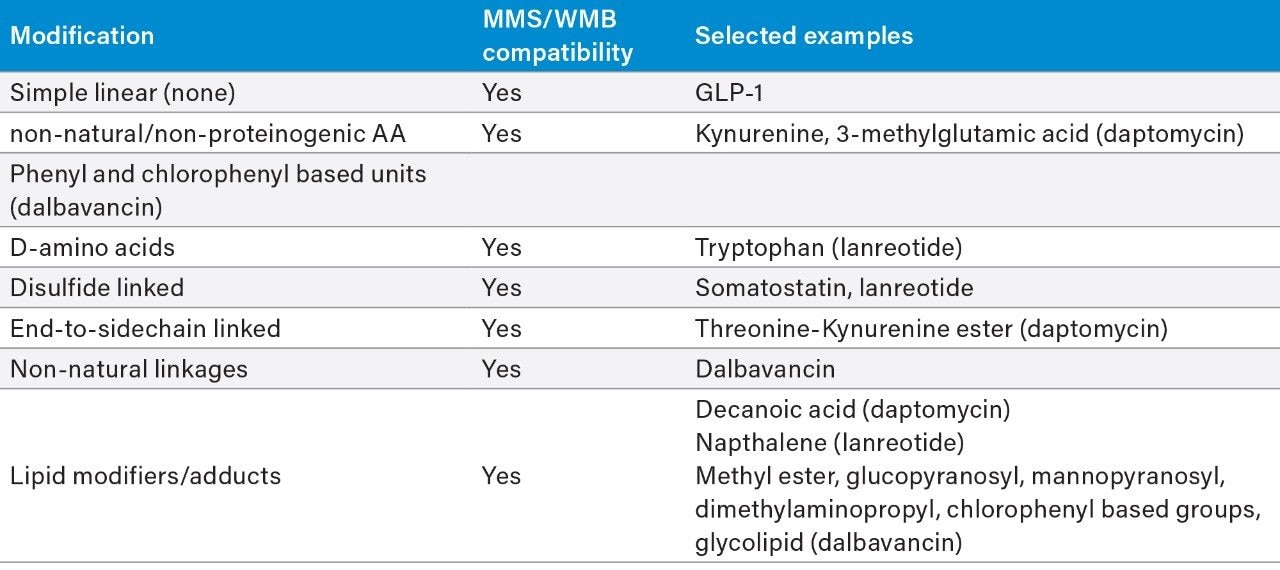
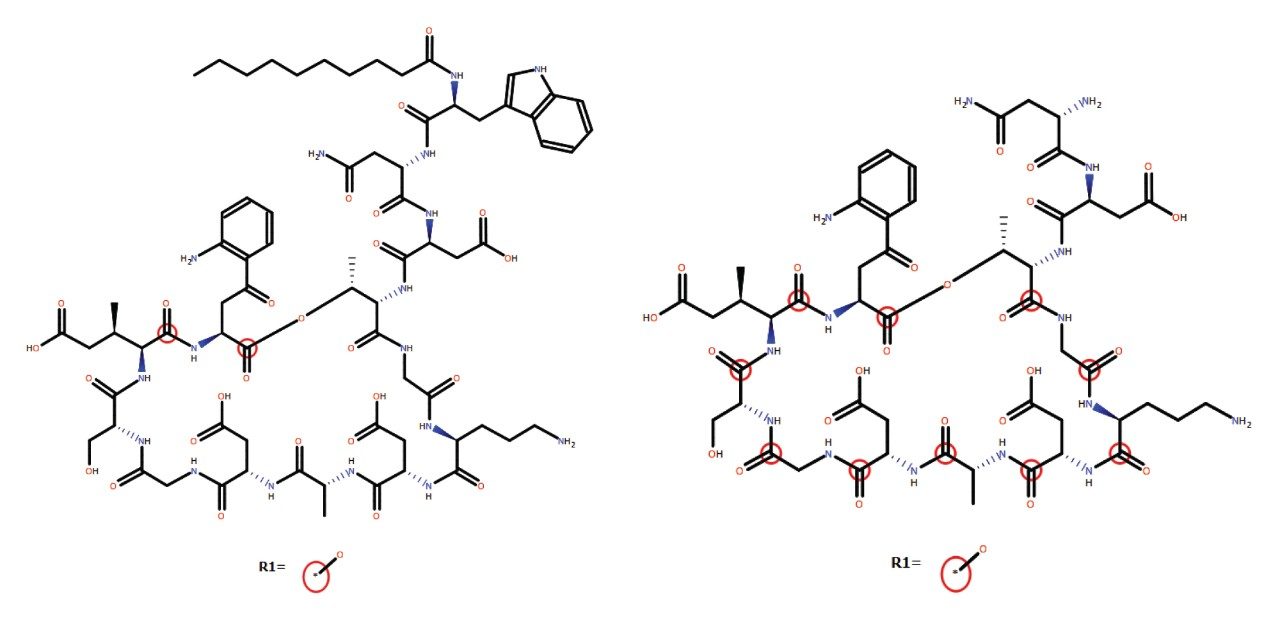
The incorporation of unnatural amino acids, backbone modifications, conjugations, and cyclizations is a commonly used strategy to improve the efficacy and ADME profile of peptide therapeutics. Analyzing these complex peptides (often natural products which may be synthetically modified) requires overcoming a variety of analytical and technical challenges, including challenging chromatography, non-specific binding, limited or complex fragmentation, and the prediction and review of many catabolites and/or novel metabolites. To ensure safe and efficacious therapies, the metabolic fate of potential drug candidates needs to be thoroughly and rapidly investigated using LC-MS coupled with informatics capable of identifying and characterizing their profiles.
Current approaches for analyzing complex modified peptides suffer from challenges that include:
- Rules developed for small molecule metabolism which are largely not applicable to or are not observed in appreciable amounts for peptides.
- Tracking and identifying all multiply charged species for all potential metabolites.
- Data Dependant Acquisition (DDA) approaches generate high quality data, but MS-MS spectra may not be triggered on relatively low abundance peaks without multiple runs, longer run times, or detailed inclusion/exclusion rules. DDA may also have an insufficient survey scan frequency to characterize all closely eluting relevant peaks. Data Independent Acquisition (DIA) (all ions) approaches are comprehensive for identification, unlikely to miss precursor information, and enable retroactive analysis, but may not always be sufficient for generating fully resolved product ion spectra for all peaks.
- Predictive approaches to generate inclusion lists can improve the quality of hits but may still miss peaks due to imperfect prediction of the precursor metabolite masses in earlier studies. Even modest peptide lengths may generate very long lists of potential targets.
- Non-specific binding, leading to underreporting and loss of some metabolites.
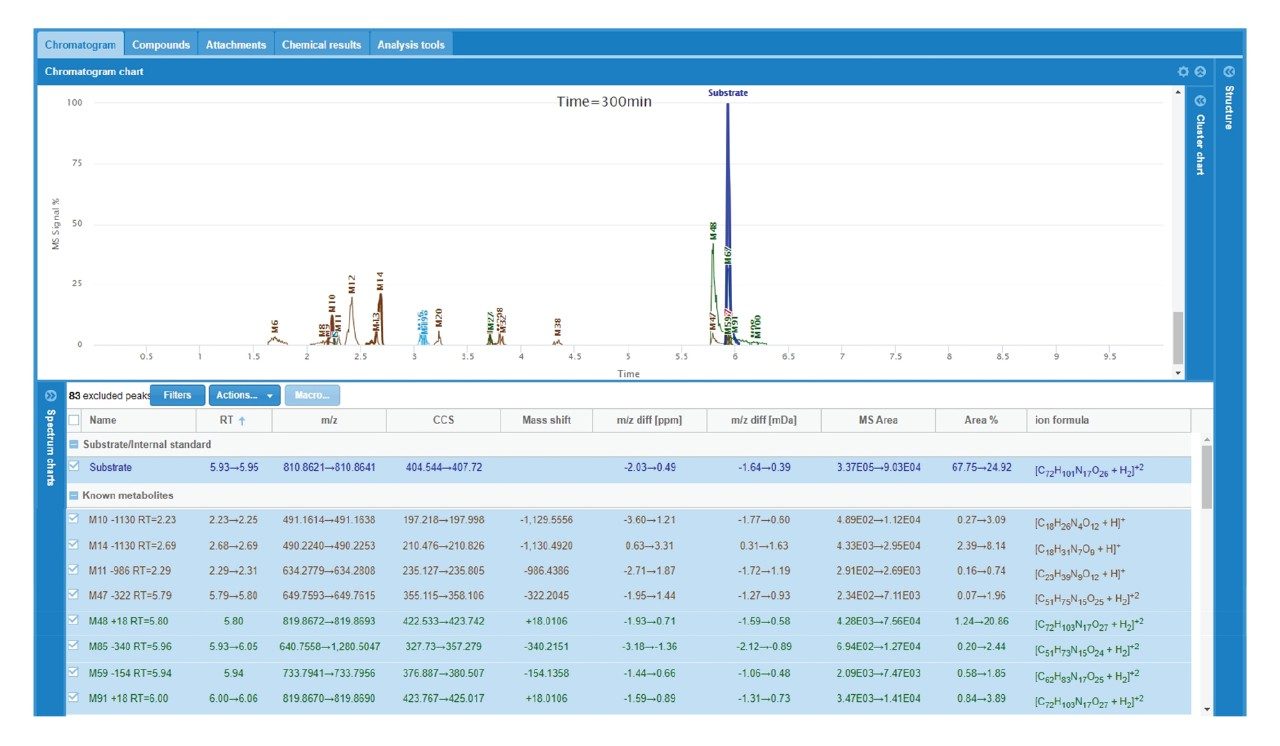
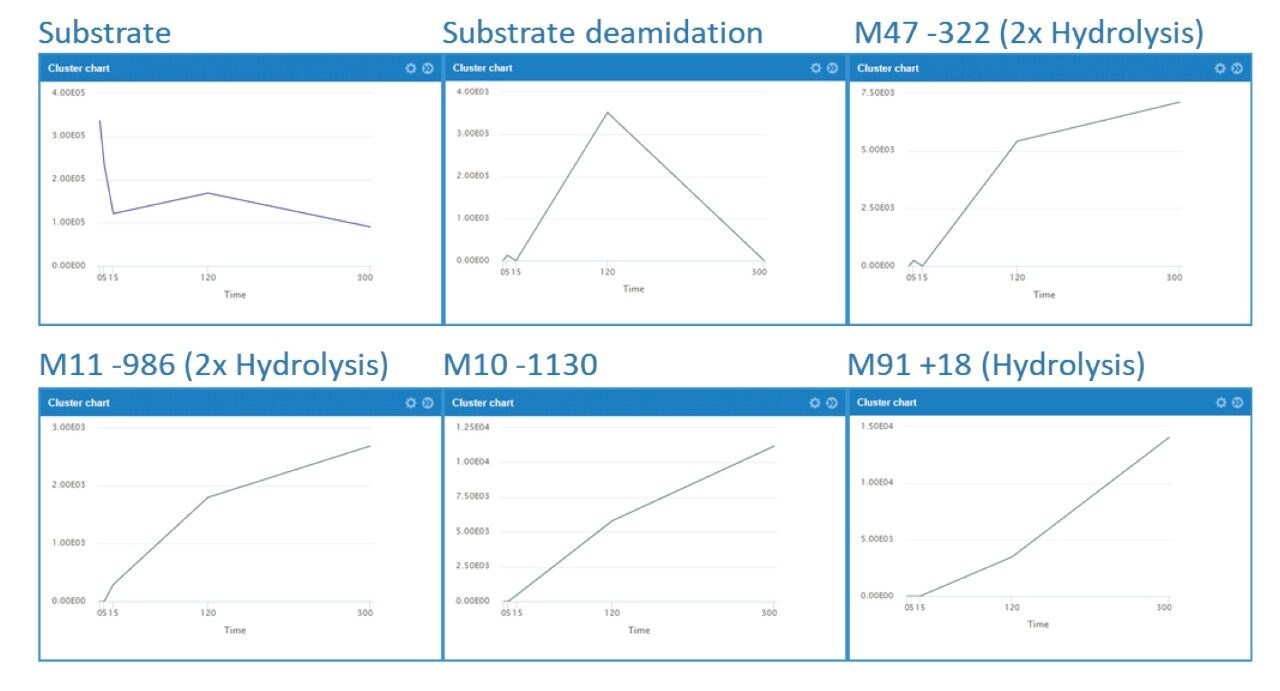
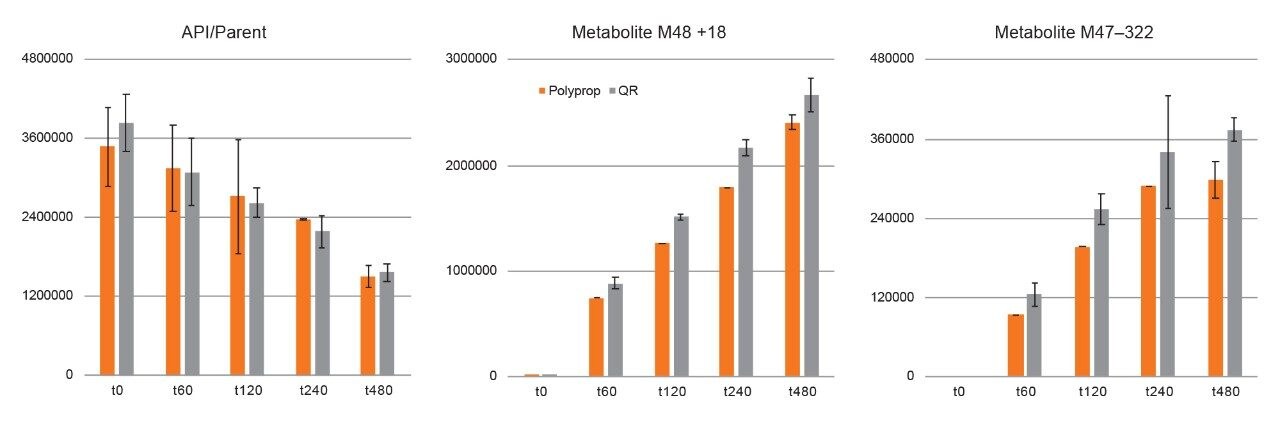
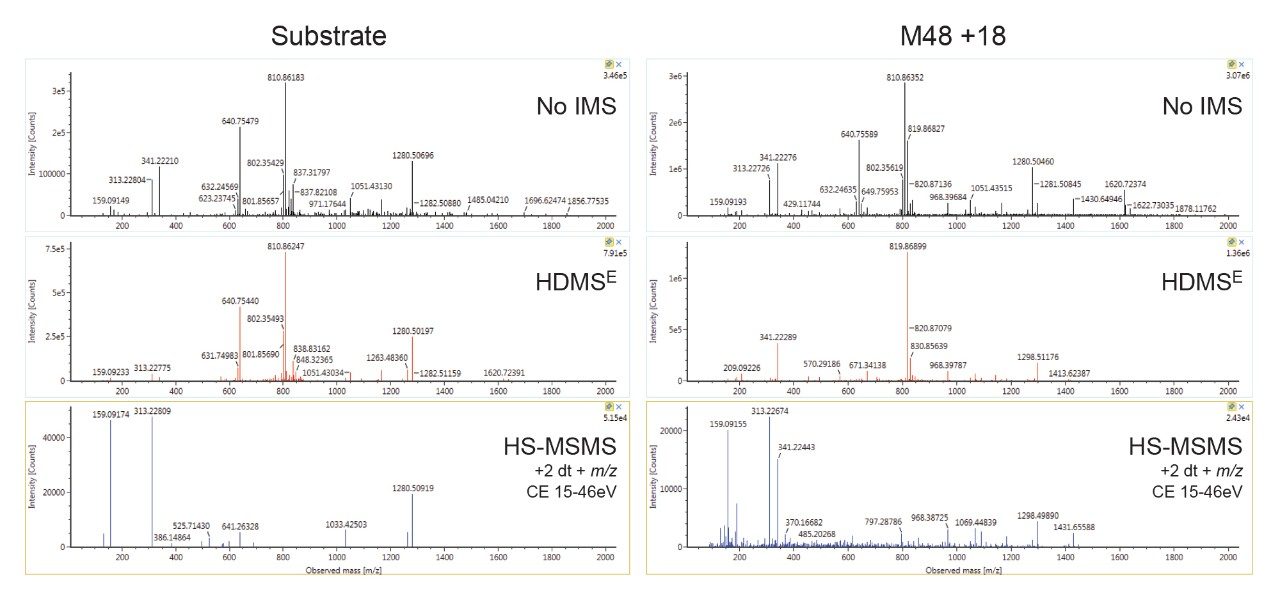
Here, we describe a solution that uses an information-rich data acquisition mode, which effectively deals with the complexity of modified peptides. This enables key metabolic data to be generated unambiguously while tackling complex peptide studies. The use of DIA combined with ion mobility (HDMSE) improves upon the selectivity provided by MS-only techniques. The addition of routine Collision Cross Section (CCS) measurements makes HDMSE a powerful identification acquisition mode, as well as an ideal separation technique. For those projects requiring definitive product ion scans, we describe the use of simultaneous product ion confirmation scans (PICS, IMS MS/MS) for metabolites with complex product ion spectra to enable the highest confidence in elucidating structures. Loss of metabolites and signal response can be mitigated through the use of low binding protocols or products such as QuanRecovery vials.
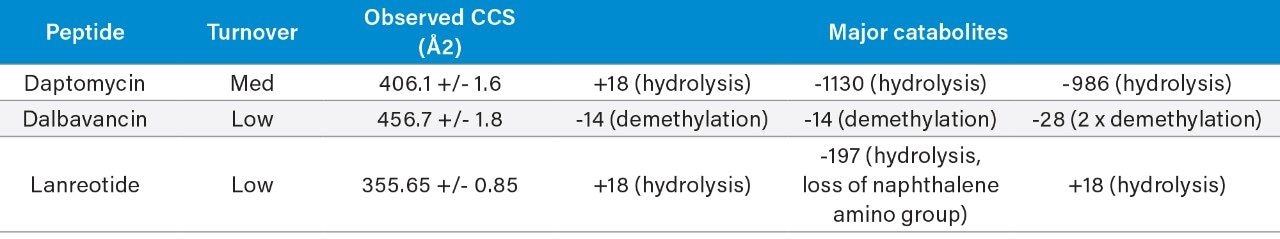
All data for daptomycin, dalbavancin, and lanreotide were screened for metabolism using an ion mobility-enabled HDMSE acquisition workflow and processed using Mass-MetaSite (MMS) and WebMetabase (WMB) macromolecule software packages (Lead Molecular Design, S.L, Sant Cugat del Vallés, Spain). HDMSE and HDMSE + product ion data were reviewed using the UNIFI screening application.