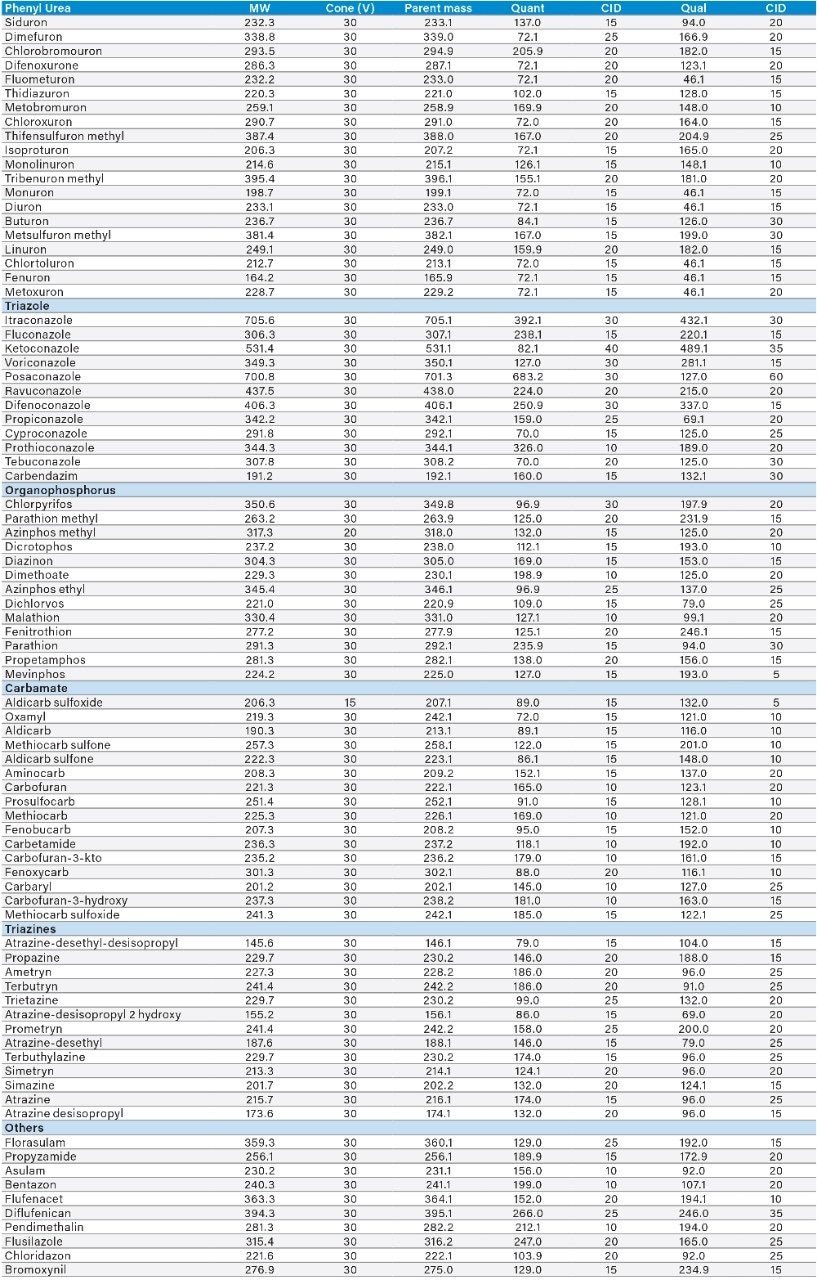
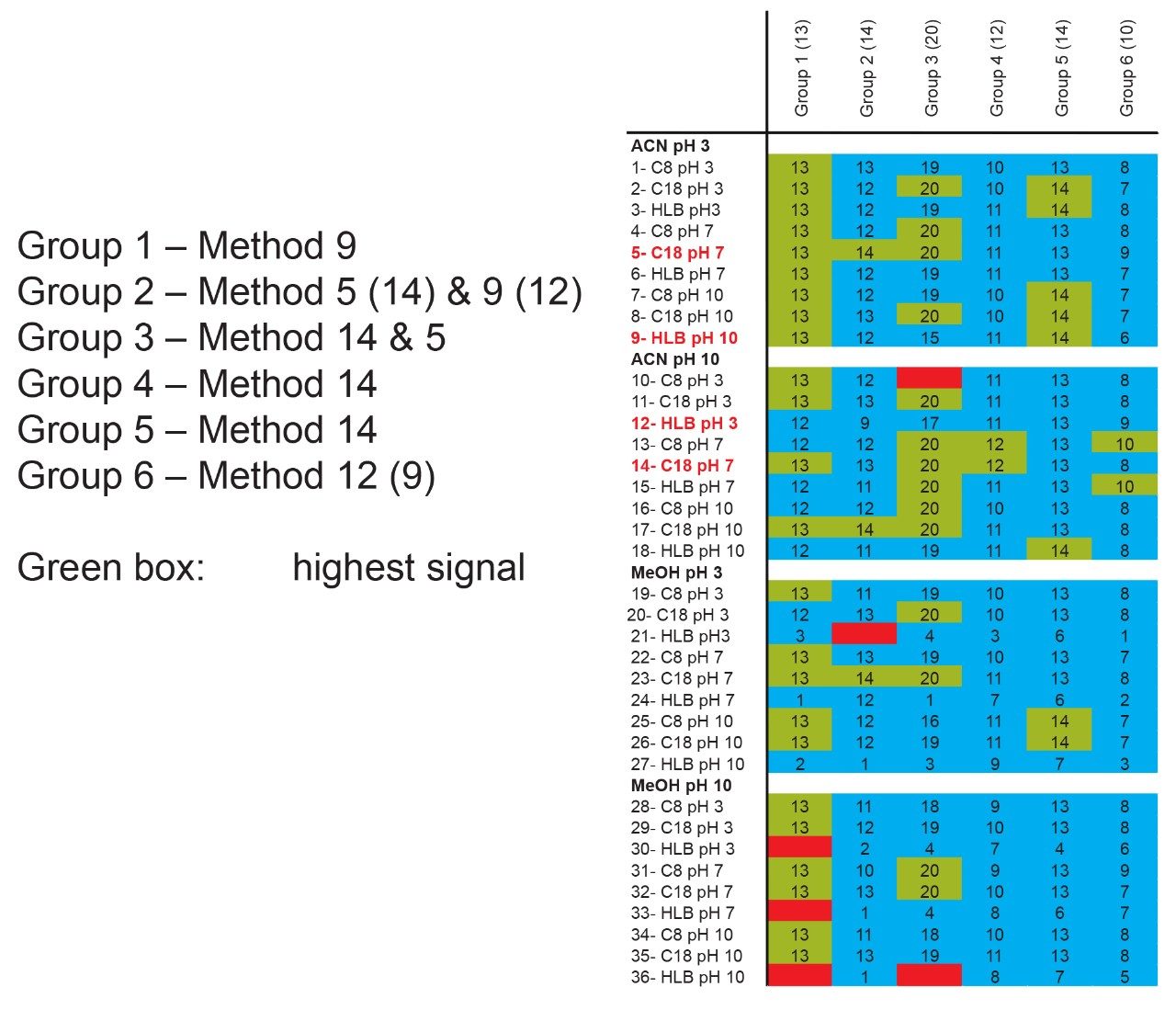
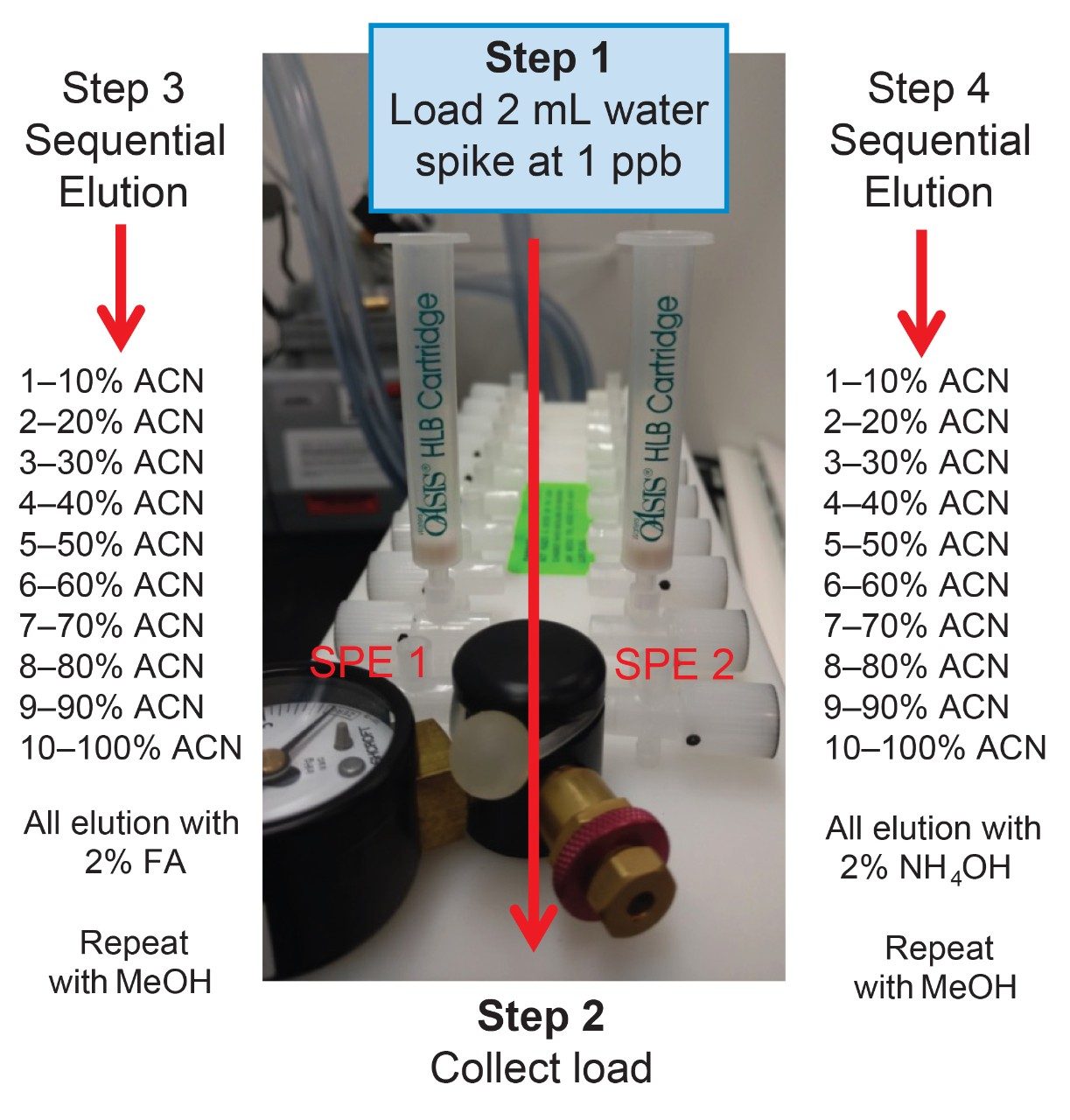
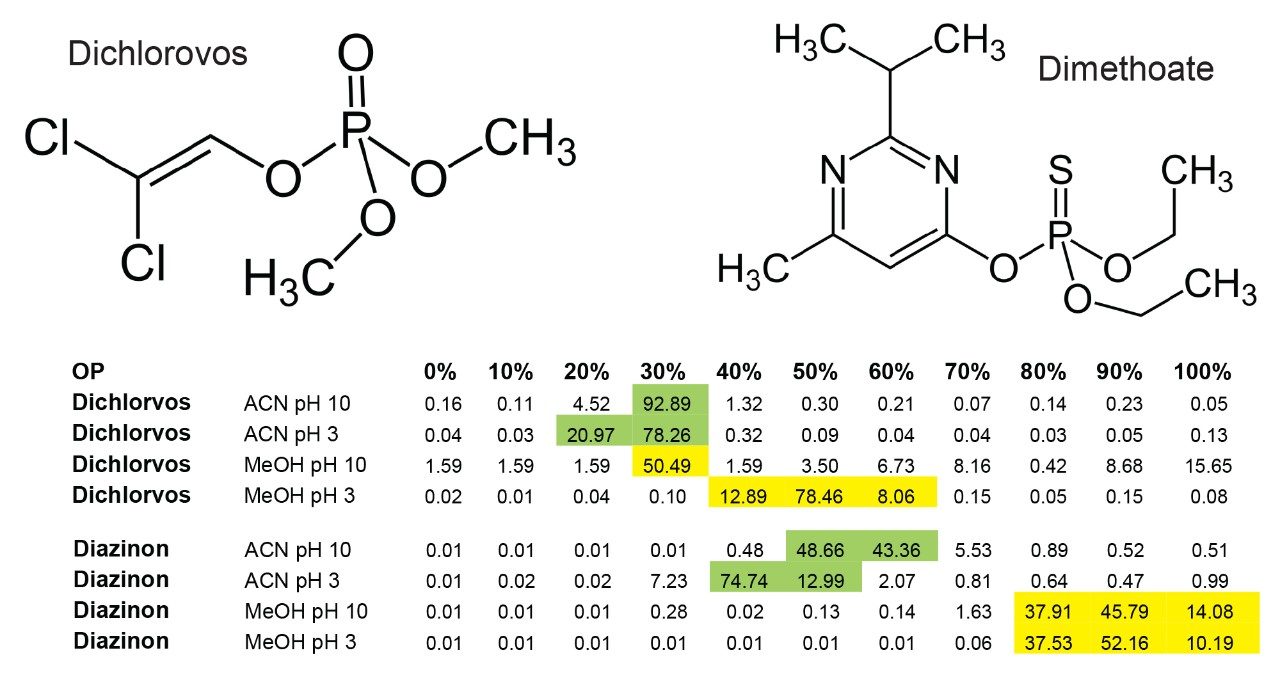
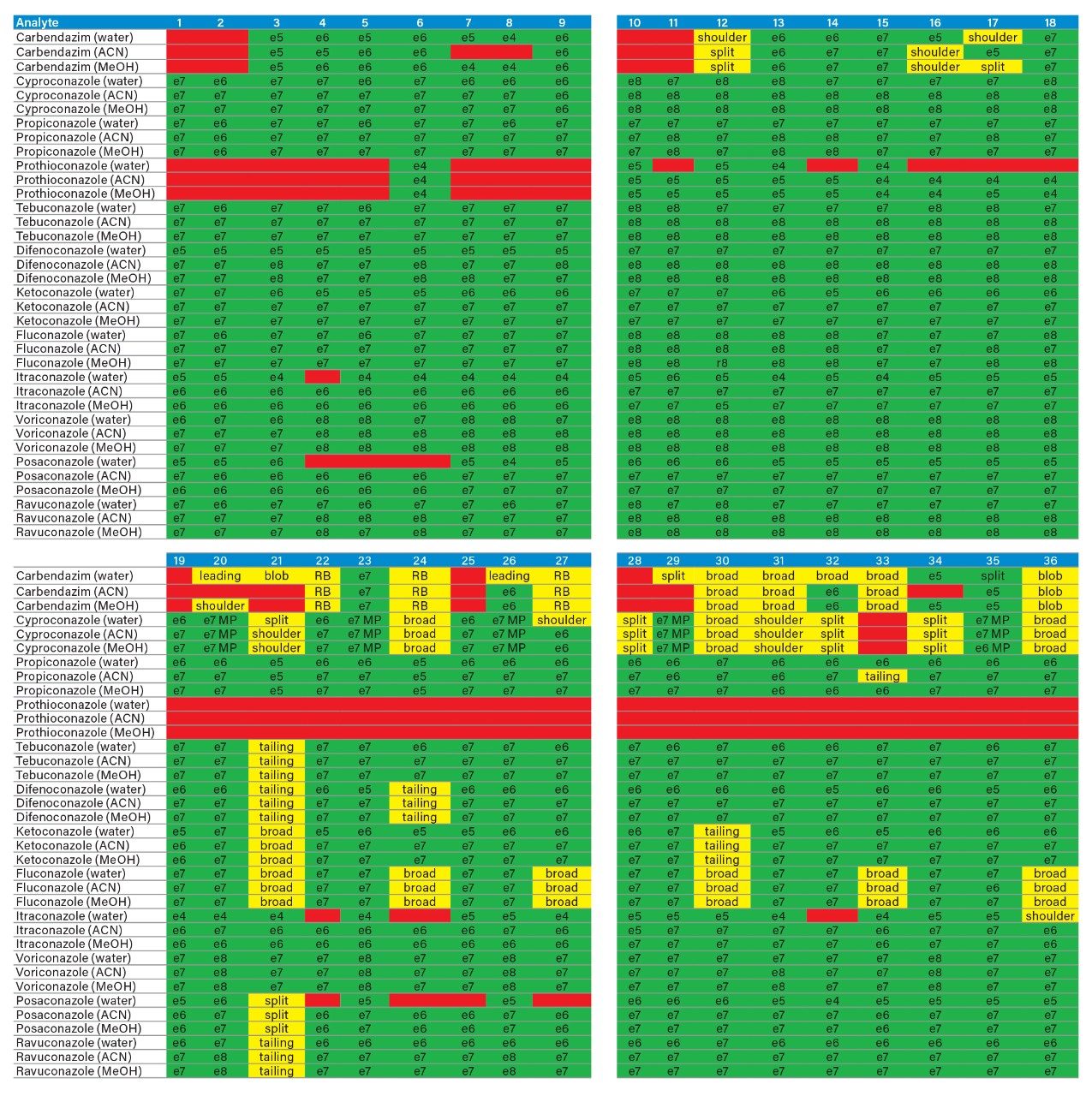
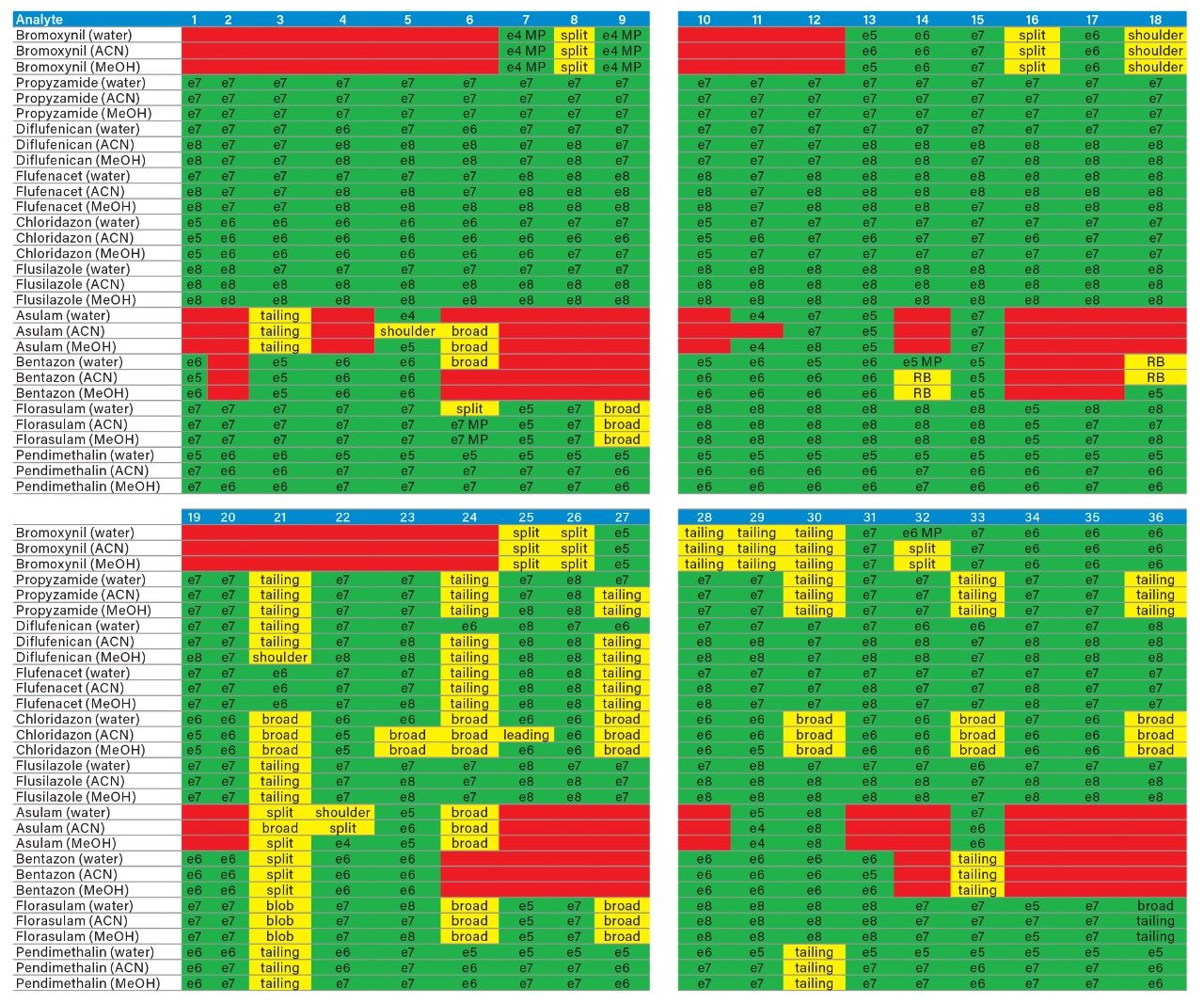
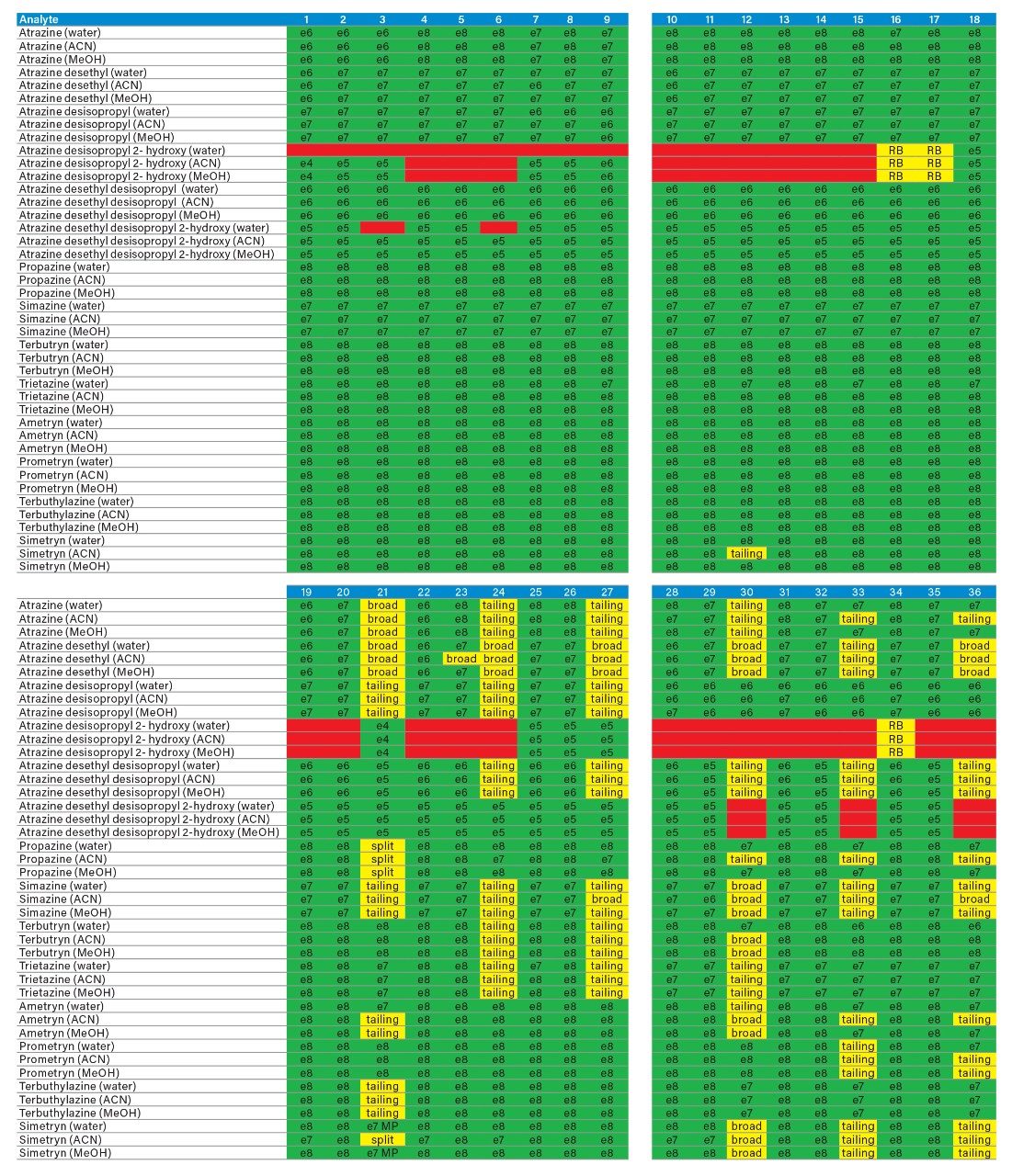
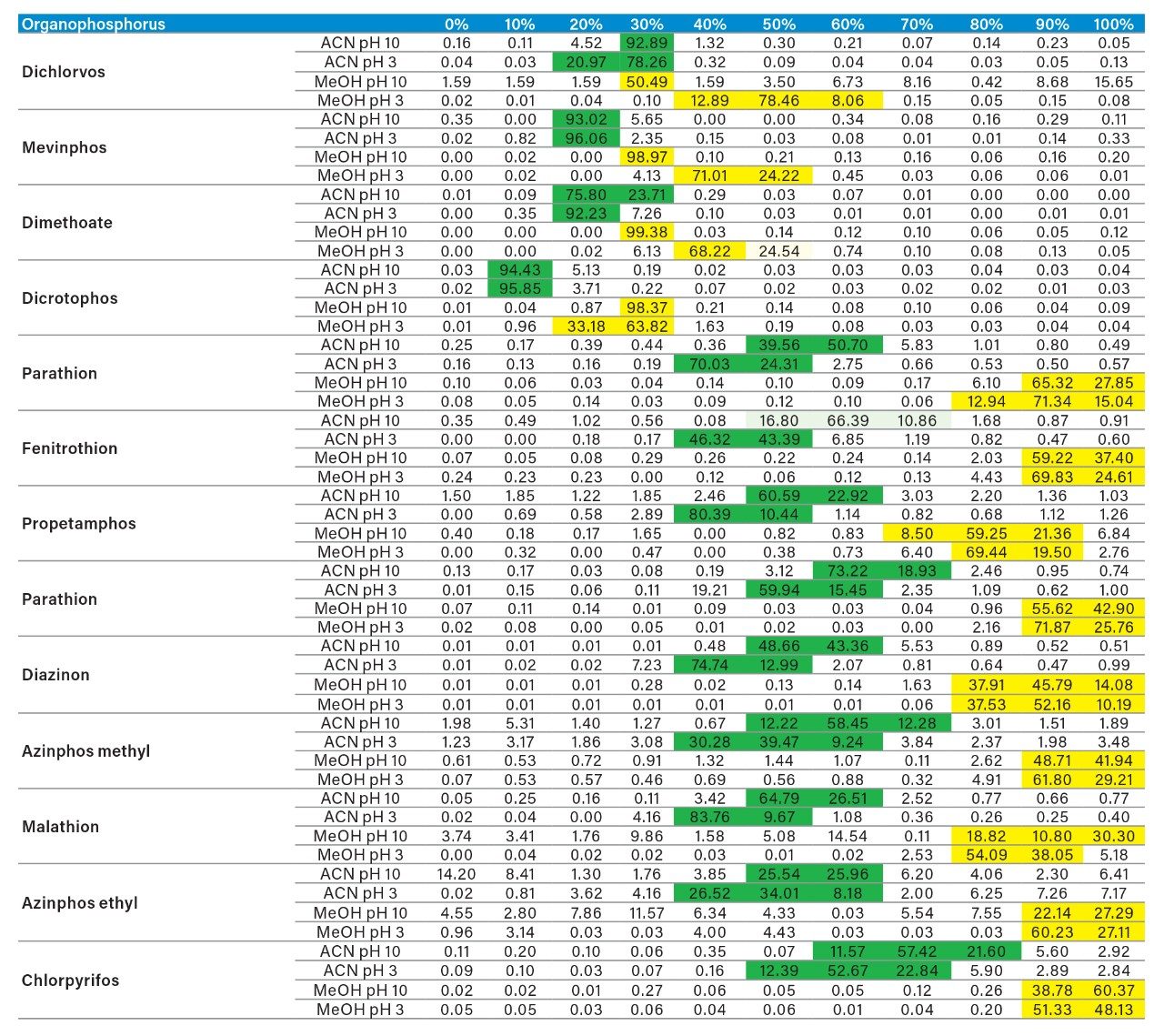
Sample preparation
The monitoring of pesticide residues in drinking water is a vital necessity for every nation worldwide. Access to clean and drinking water quality is the EPA primary focus in United State and began with the Safe Drinking Water Act in 1974. Pesticides analysis in drinking water sample is a time and laborious process, with the bulk of the work consumed by lengthy sample preparation protocols. In most instances, due to low detection requirement (sub ppb levels), high enrichment extraction protocol must be used to bring target pesticides concentration level in a sample up to detection level compatible with current separation and detection techniques. As such, most extraction methods will incorporate up to 1000:1 enrichment factor to add up to 1000-fold sensitivity increase for quantification. In practical terms, large volumes of a raw sample must be concentrated to an extreme low volume; the most popular method will utilize a 1000 mL starting sample volume to a final extract of 1 mL, thus providing the necessary 1000:1 enrichment factor. Several extraction methods can be used, liquid-liquid and solid-phase extraction are amongst the most utilized for this type of application. Regarding SPE, this technique offers a wider performance than liquid-liquid extraction; in most applications SPE is the favoured method but not without difficulties. With SPE, once a sorbent bed is conditioned, a target volume of water sample will be loaded to ensure total and maximum contact with solid particles for extraction of target analyte. This step is crucial and requires a minimum flow rate per bed mass. With regards to high enrichment extraction, SPE bed mass in the 50 mg and higher range are the most common and avoids breakthrough effects due to saturation with smaller bed size. With the SPE barrel format, coarse filters are used for the top and bottom to ensure that the bed density remains compact. With those facts in mind, the loading flow rate is usually set between 5 to 10 mL/min and can be set with proper valve setting on a vacuum manifold. For a typical 1000 mL drinking water sample loaded at 10 mL/min, it will take roughly two hours for the entire sample to interact with the sorbent bed.
However, if the water sample contains high level of organic matter (Total Organic Content or TOC), the top filter fritz will collect all particulate in the sample and will cause a restriction effect over time, meaning that the loading flow rate will drop at a lower value. With increasing level of TOC’s, the first 100 mL of the sample is usually loaded at optimum flow rate but will rapidly drop to extreme low values and increases the loading time. For example, a tap water sample can take up to 8 hours of loading time, and up to 15 hours for a surface water sample. With a typical lengthy extraction process associated with high enrichment protocol, it inevitably led to the creation of multi-residue applications.
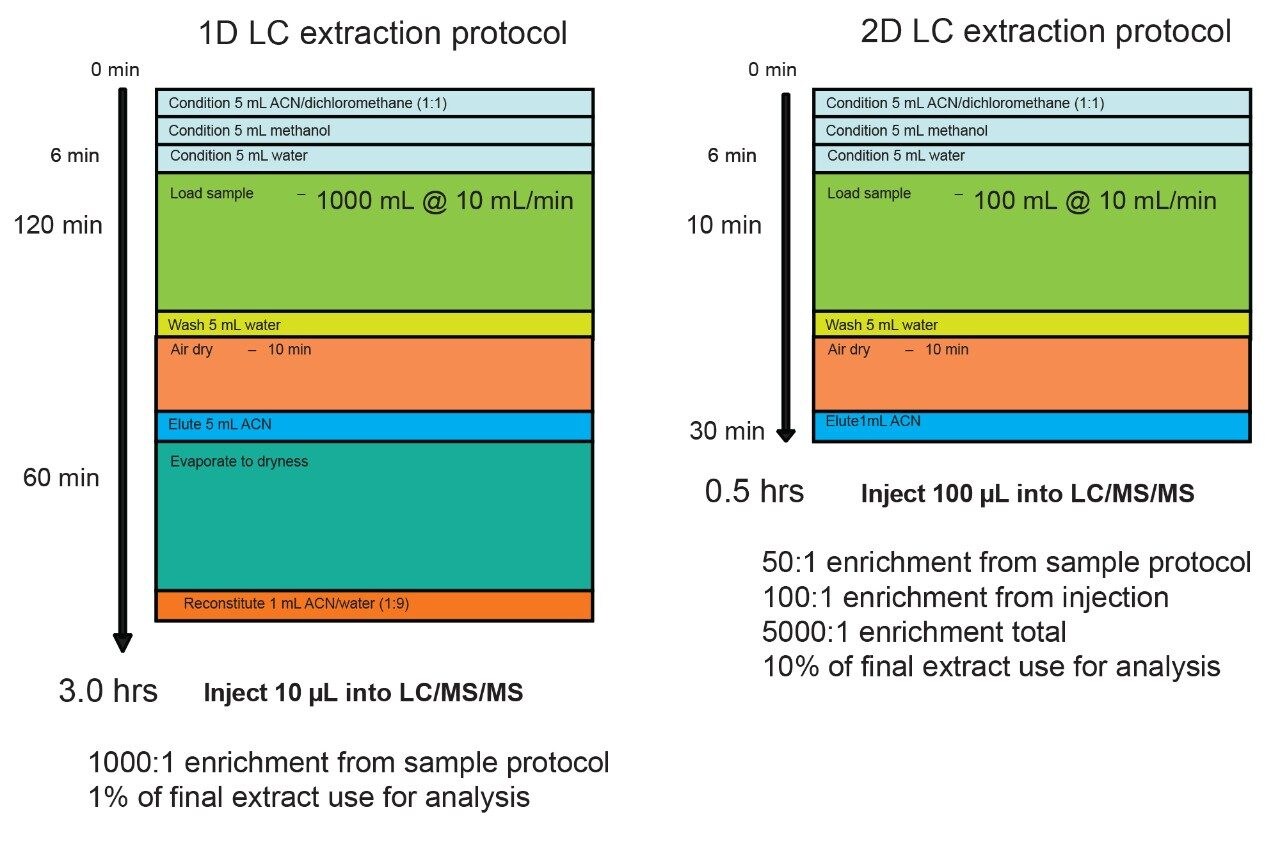
This type of enrichment protocol is linked to the limitation of single dimensional separation technique. The low volume injection restriction (10 µL) is a necessity to avoid volume or mass overload during the injection sequence when combined with a single dimension chromatography platform. Recent development in multi-dimensional chromatography offers the option to create customizable configurations for a specific workflow, thus leading to the possibility to create novel extraction protocols. The 2D LC with At-column dilution configuration offers the option of large volume injection without the distorted peak shape effect due to volume overload. This particular configuration also offers the particularity to inject aqueous and organic solvent extracts at any percentage. Previous works describes the technique in detail. Ultimately, a water extraction protocol using a 2D LC-MS/MS platform is completed in minutes instead of hours. Since a large volume can be injected (up to 1000 µL) on column, the loading sample volume during the extraction process can also be reduced to strict minimum. For example, using a 20 mL sample volume with a final elution at 2 mL can lead to a 500 uL injection of a 100% organic extract, thus producing the same 1000:1 enrichment (See Figure 1). The added benefit is the 2 mL loading time and the elimination of the dry down step, in most application the evaporation with nitrogen stream is simply eliminated from the protocol. If higher enrichment is required, up to 200 mL of sample can be use and while maintaining the total extraction time below 30 minutes.