Recovery
Determination of oleic acid content (free and esterified) has been shown to be used successfully in the assessment and extrapolation of PS-80 degradation and concentration in drug products.2 More recently oleic acid has been used successfully as a predictor for functionality related characteristics such as critical micelle concentration.8 Analyses such as these often employ liquid-liquid extraction (LLE) to isolate fatty acids from sample matrices. However, LLE techniques are not without their own challenges such as matrix effects, extraction volume, and solvent type, which can negatively impact recovery efficiencies and impair accurate quantification. Furthermore, organic extraction solvents are often immiscible with aqueous buffers resulting in laborious dry-down and reconstitution steps. These challenges are addressed in the current study with the development of an extraction protocol that can be used for the extraction of free and esterified fatty acids (after hydrolysis) and is amendable for direct injection onto columns with minimal sample preparation.
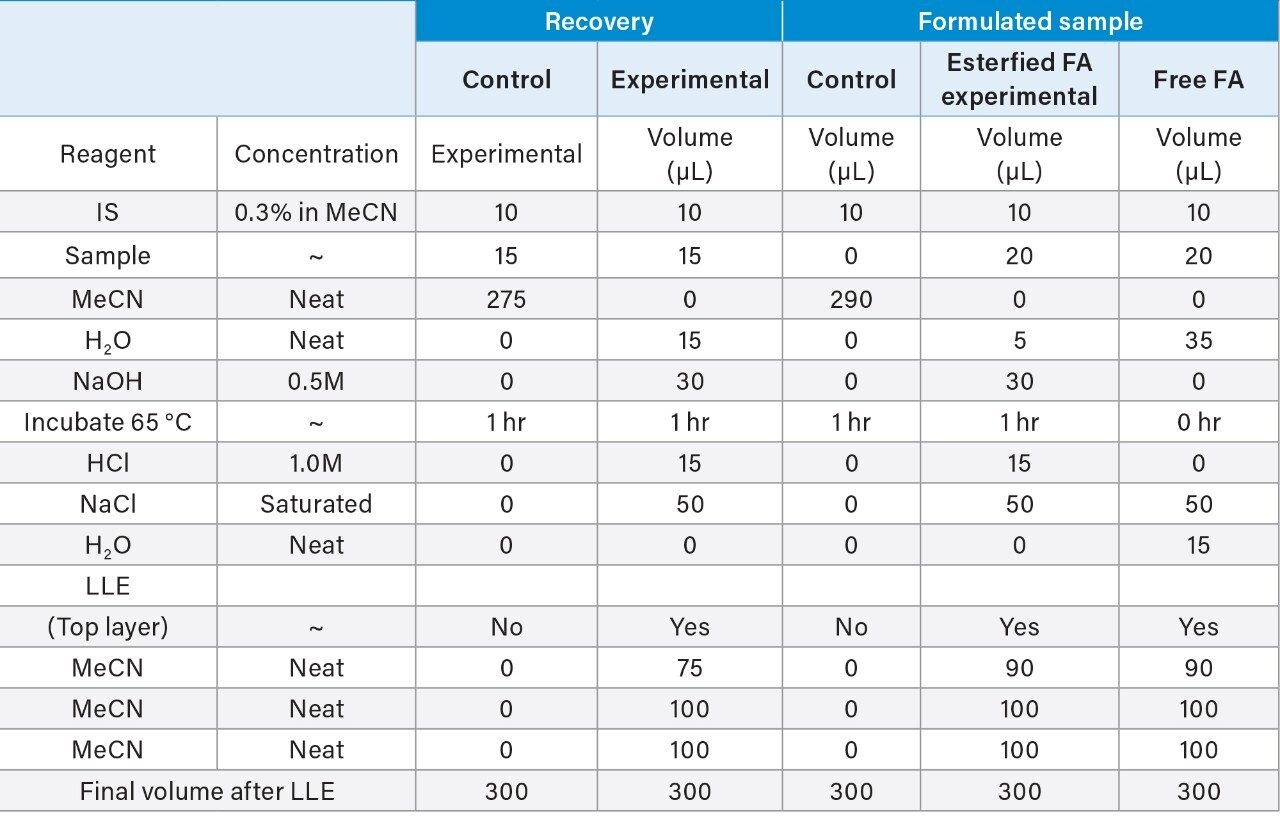
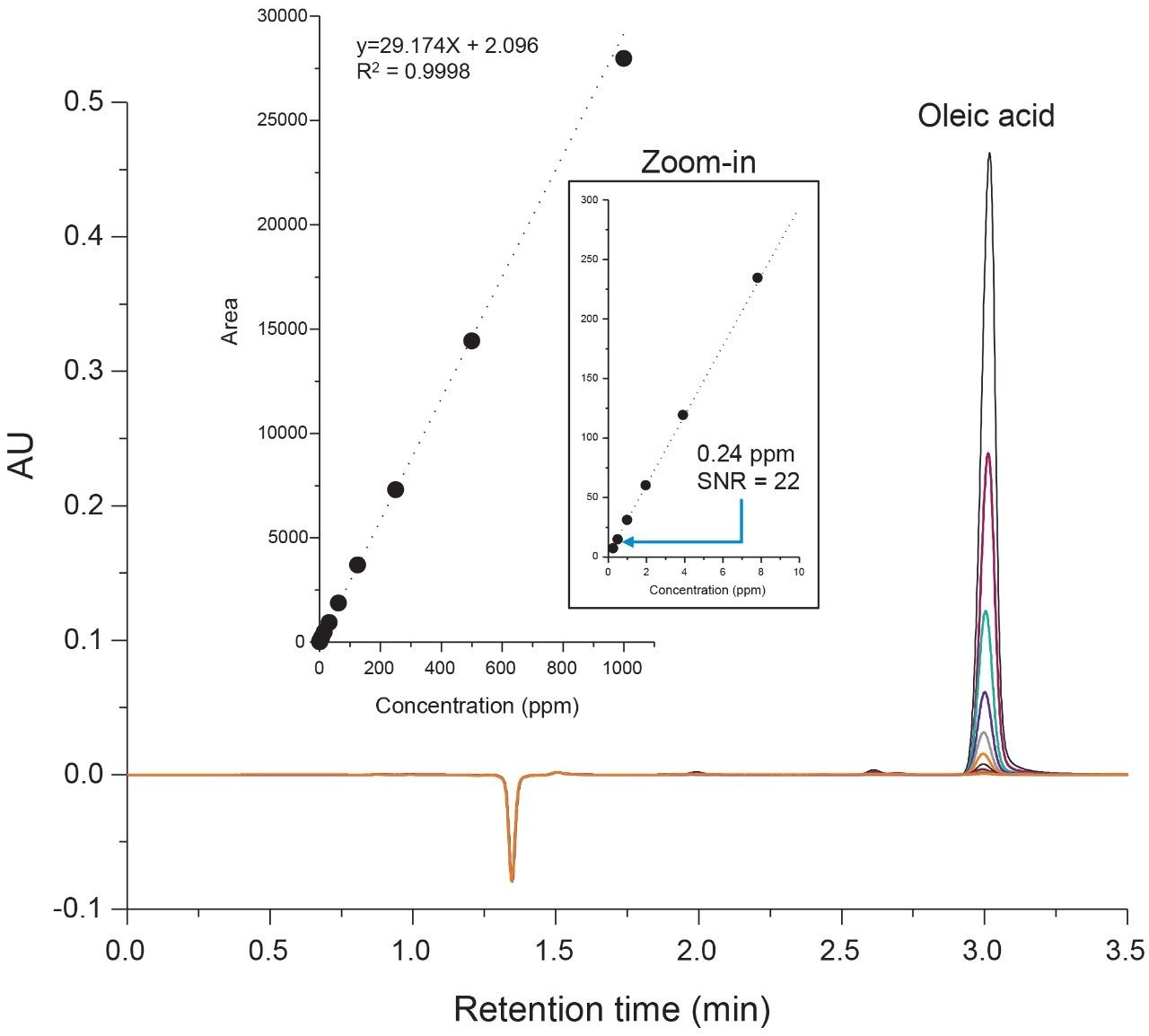
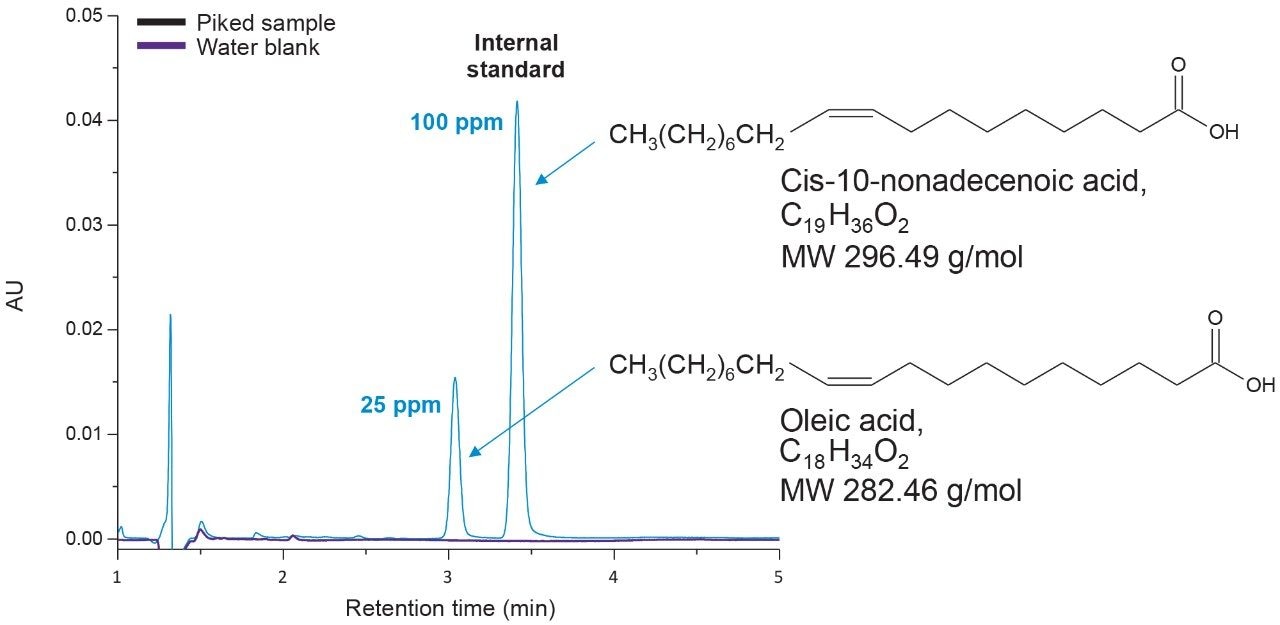
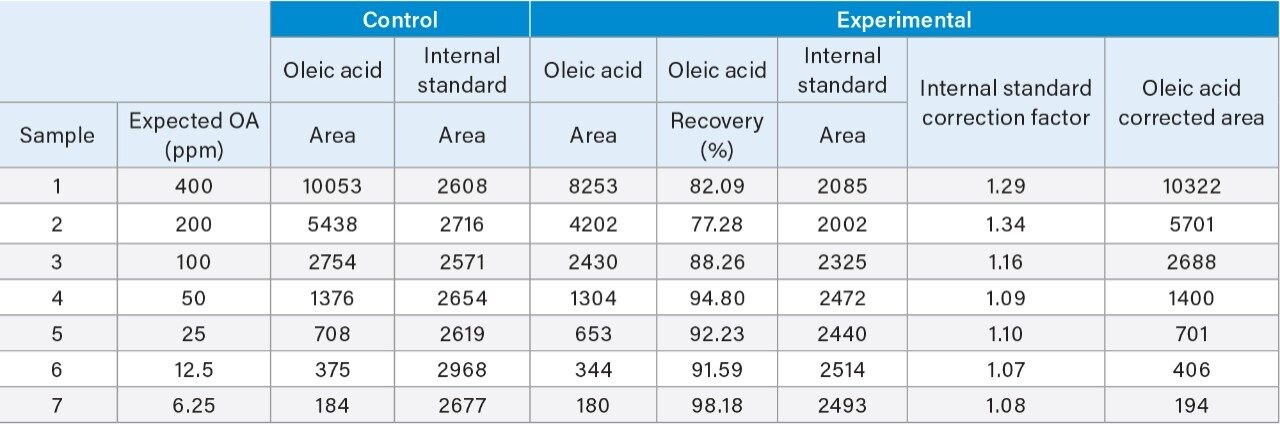
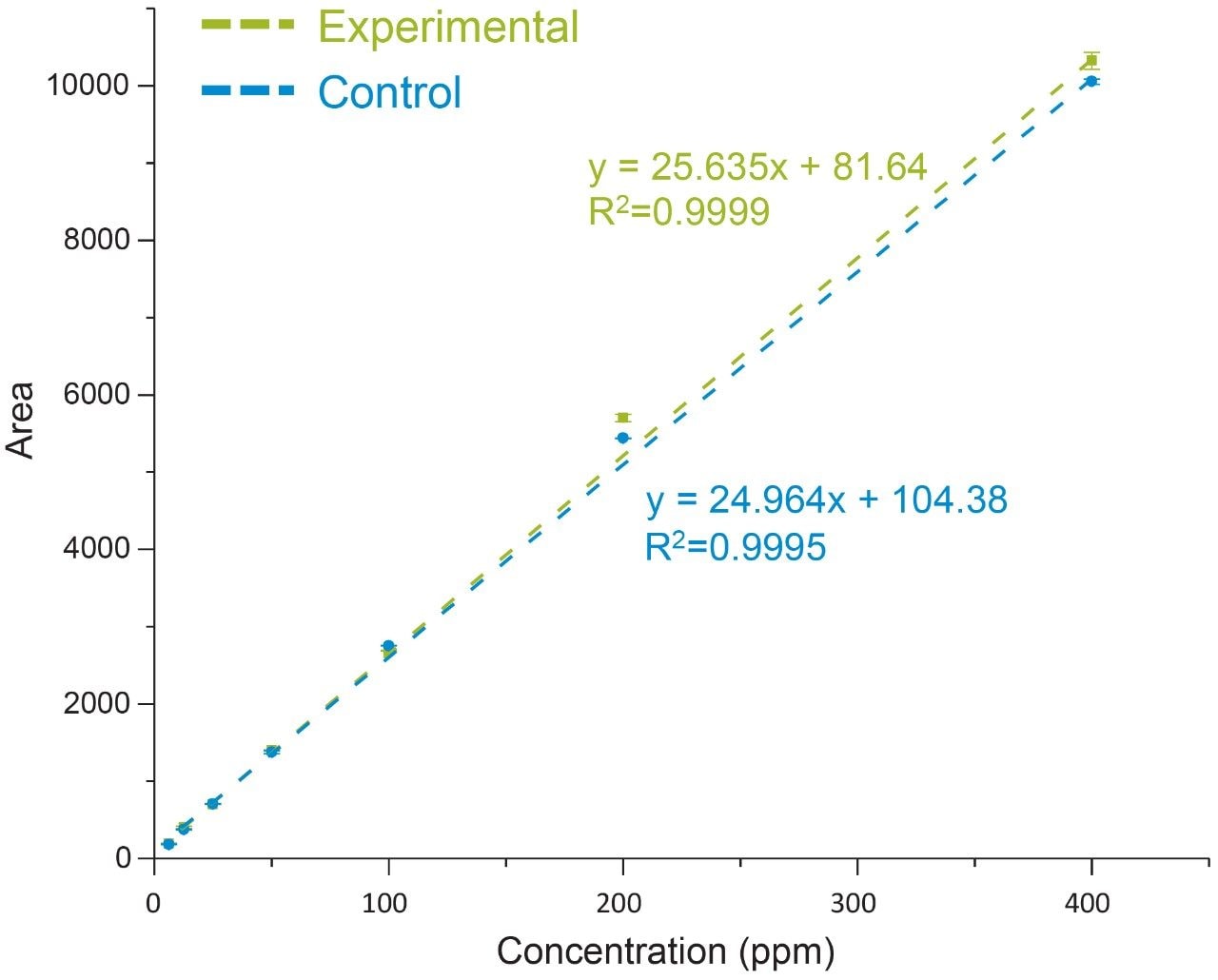
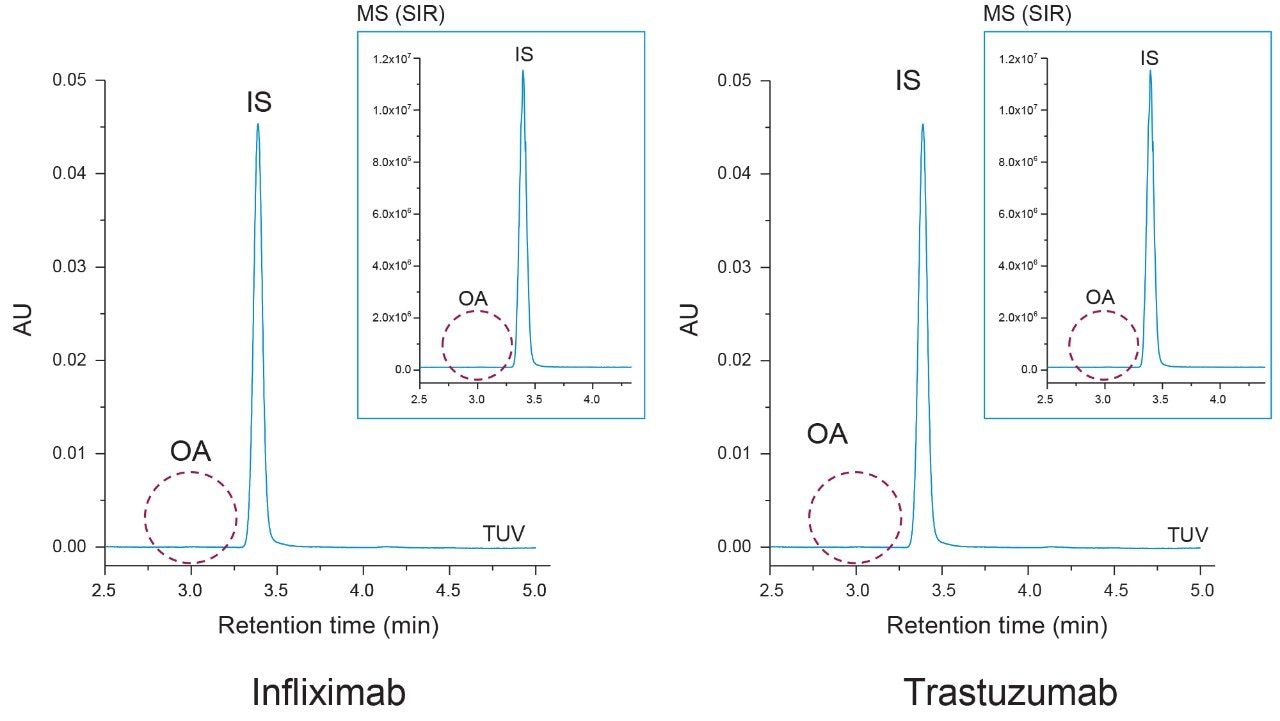
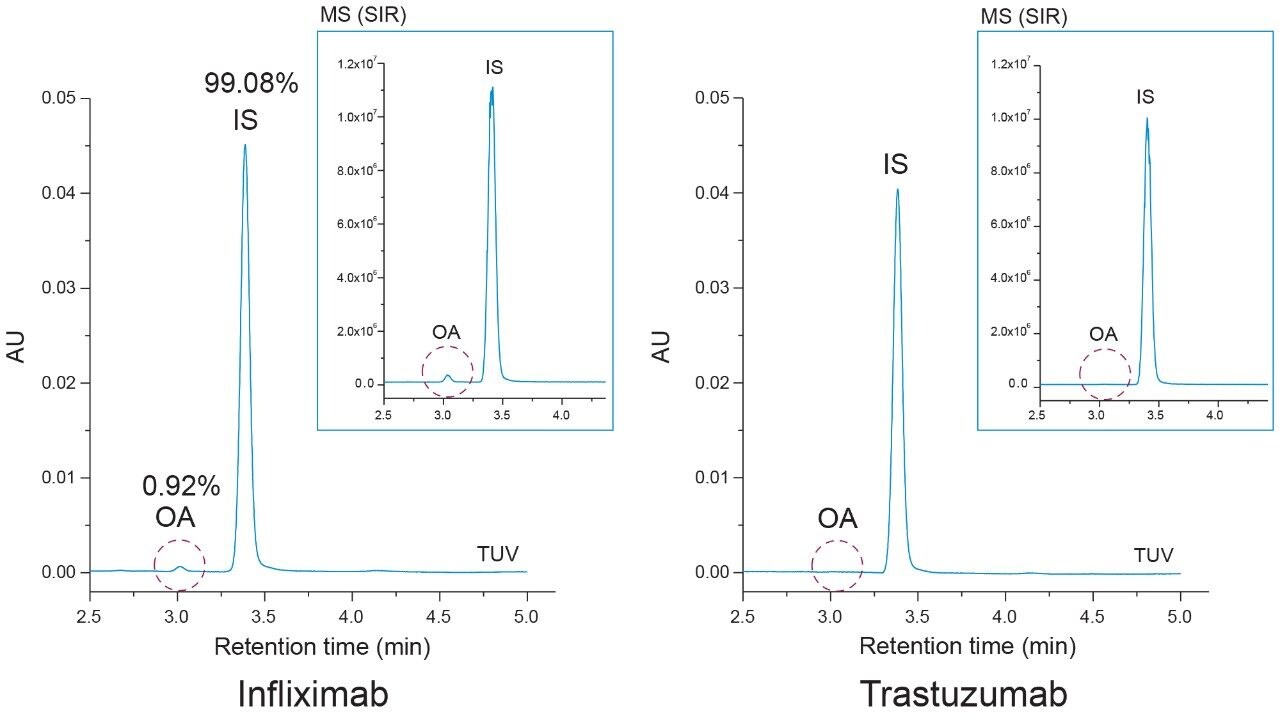
The extraction protocol was evaluated for stability, recovery efficiency, and column performance prior to drug product analysis using an oleic acid standard. Cis-10-nonadecenoic acid was used as an internal standard to correct for non-specific loss of oleic acid during the extraction process. Two sets of oleic acid samples representing experimental and control were prepared as outlined in the extraction protocol (experimental section). Briefly, stock samples of oleic acid were prepared in MeCN at concentrations 0.8%, 0.4%, 0.2%, 0.1%, 0.05%, 0.025%, and 0.0125%. Experimental samples were prepared by taking 15 µL aliquots of each stock and spiking in 10 µL of the IS. NaOH was added to the experimental set and incubated at 65 °C for 1-hr to assess oleic acids stability at the elevated temperature and high pH required for base hydrolysis of PS-80 and drug product biological matrices. The solution was then acidified with HCl to increase solubility of residual biological material in the aqueous phase as well as minimize the column exposure to elevated pH that could decrease column longevity. After acidification, a saturated solution of NaCl was added to induce a phase separation between the acetonitrile and aqueous phase.3 Initial extraction of the top-layer was performed with a reduced aliquot of MeCN to account for the MeCN present in the oleic acid sample and internal standard. A control set was prepared in MeCN to determine recovery efficiency and correct for non-specific sample loss. As shown in Figure 3, a direct injection of 1 µL of the extracted spiked in sample using the same isocratic conditions as before is sufficient to separate oleic acid from the IS with baseline resolution. A water blank (blue trace) performed after the injection showed no observable carry-over on the column. Recovery efficiencies up to 98% were observed for samples containing trace amounts of oleic acid as shown in Table 1 with a marginal decrease in recovery efficiency observed at higher concentrations. Sample loss was determined to be non-specific, as similar trends were observed with the IS as well.