According to the Scientific Working Group for Forensic Toxicology (SWGTOX), the field of forensic toxicology handles the analysis of drugs or chemicals in biological materials, and the interpretation of those results for medico-legal purposes.1 In this field, forensic toxicologists often work with medical examiners to perform postmortem toxicological analyses on blood or biological tissues of deceased individuals in order to determine cause and manner of death.1 Because these results are relied upon in a court of law, validity, reliability, accuracy and precision of the analytical techniques used to perform these analyses are essential.
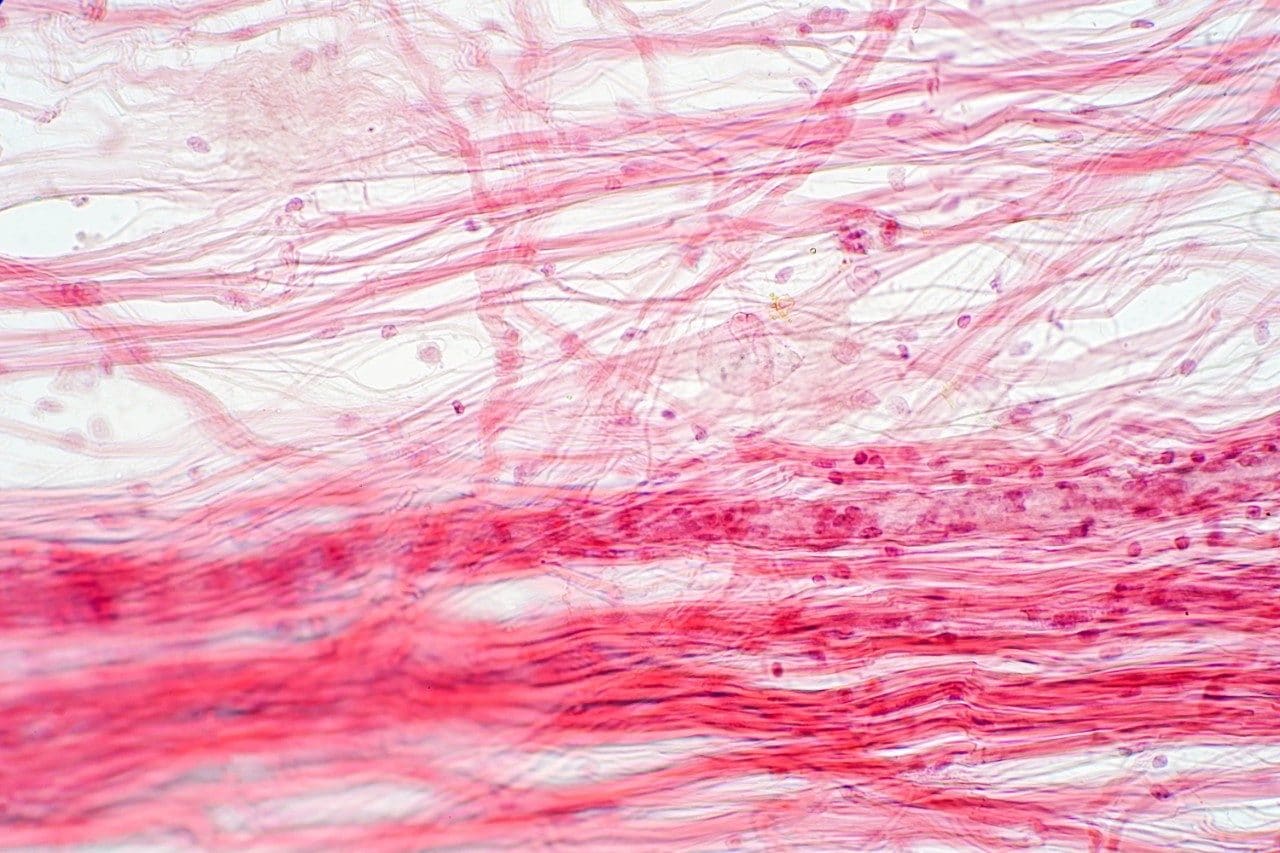
The core focus of a forensic toxicology laboratory is the accurate identification and quantitation suspected drugs or chemicals in biological samples. The target matrix can vary between blood, plasma, urine, saliva, vitreous fluid, hair, nails, and organs such as brain, heart, lung, liver, kidney, spleen, and stomach contents. The Forensic Toxicology Research Team at the Federal Aviation Administration performs such analyses on samples from victims of fatal aircraft accidents to provide insight to the analysis of accident causation.2 Aircraft accidents and crashes are often brutal enough to severely impair any human remains, which is why the toxicologists must rely on more complex biological tissues for analysis, i.e. brain, heart, lung, liver, kidney, spleen, etc. Additionally, they must have the ability to detect and measure many substances, from drugs and alcohol, to toxic gases and industrial chemicals.2 Therefore, there is a need to develop multi-residue analyses and efficient sample preparation methods in order to analyze samples in a timely manner.
The analytical techniques currently available are divided into two categories, some platforms are used for screening methods (qualitative) and other solutions are used for confirmation methods (quantitative). Most laboratories are usually equipped with gas chromatography (GC) or liquid chromatography (LC) hyphenated to a mass spectrometer (MS). For several decades, GC-MS was the tool of choice for bio-analysis. With the introduction of atmospheric pressure ionization technique, LC-MS is now the most popular technique in the field of forensic toxicology.
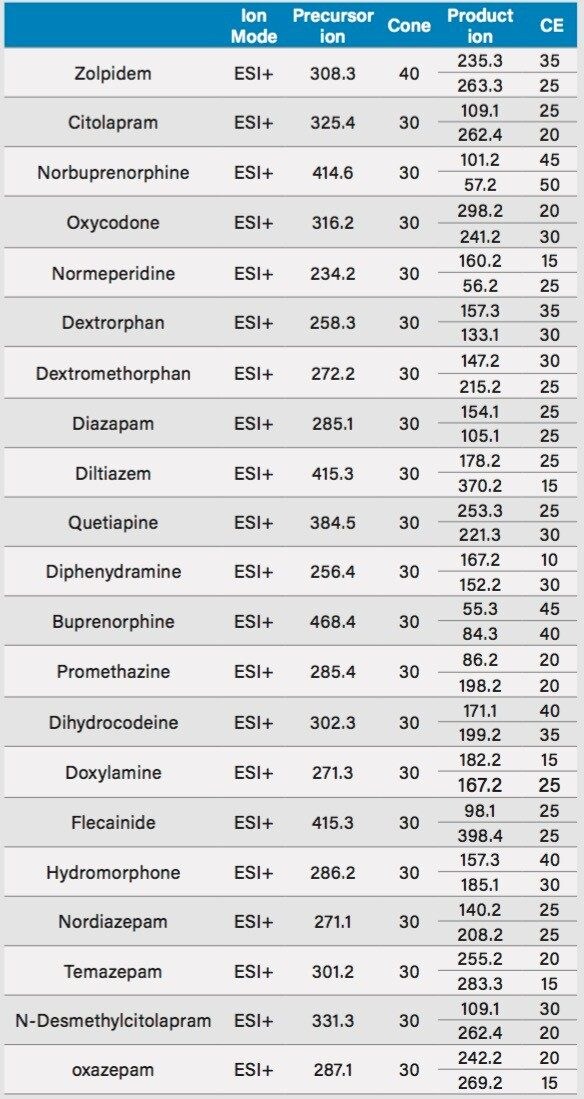
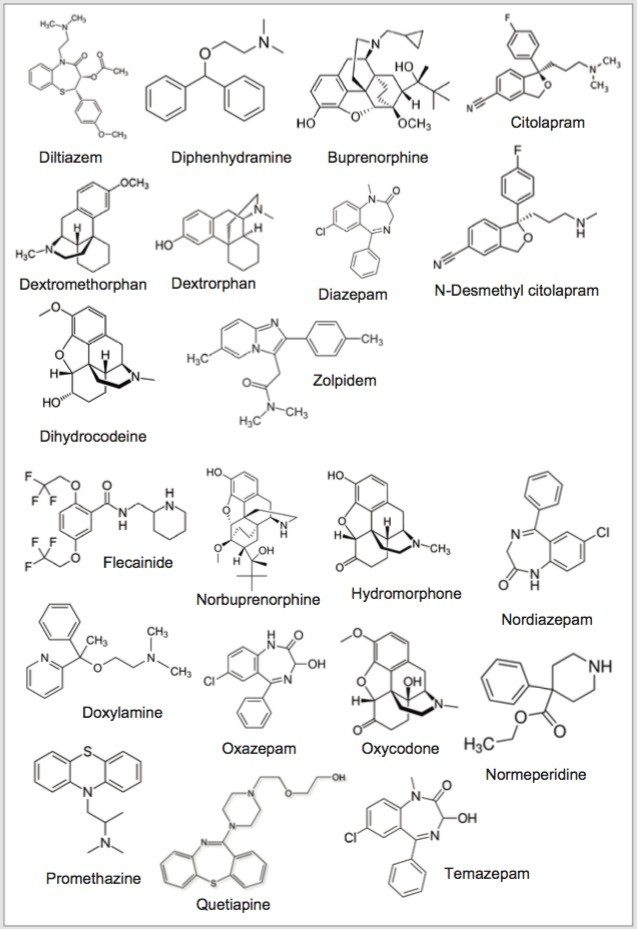
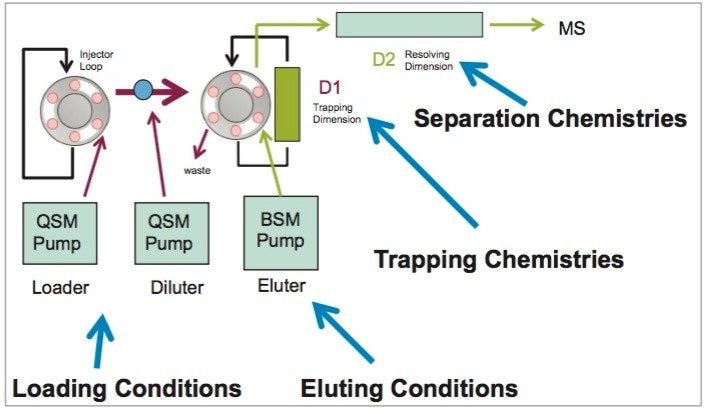
Detection and quantification of drugs in complex matrices is difficult to accomplish due to time-consuming extraction processes, and the difficulty to detect an analyte at trace levels. A robust extraction and clean up methodology, in which a homogenization step precedes, is a must in order to reach a target limit of detection (LOD) and to maintain instrument performance. The use of advanced hyphenated instrumentation platforms, such as UPLC-MS/MS has allowed analysts to detect trace levels of analytes. Traditional extraction techniques used in most laboratories are decades old and do not have the robustness to produce quality results. A micro extraction protocol combined with a multi-dimensional chromatography (2D LC-MS/MS) can decrease sample preparation time without sacrificing the quality seen with current single dimension chromatography techniques.3,4,5