Sample and Instrument Preparation
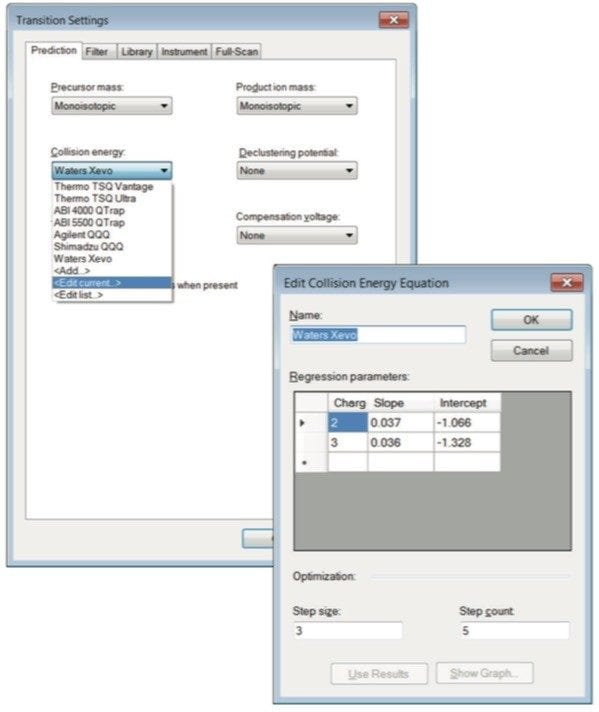
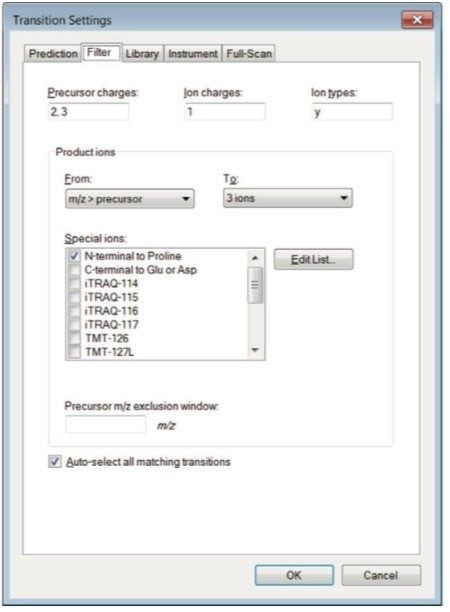
Step 1. Configure Skyline for Waters mass spectrometers by changing the collision energy to “Waters Xevo” in Settings > Transition Settings > Prediction tab (Figure 2). Additionally, the number of collision energy steps and intervals to investigate can be modified by clicking on ”Edit current” under “Collision energy.” The optimization ”step size” can be set to 2 and the ”step count” 5. Step count is the number of different collision energies to try above and below a predicted value, and step size is the difference between those collision energies. Under the Filter tab, change ”precursor charges” to 2, 3. The “ion charges” (charges on the product ions) value could be 1 or 1,2 –useful for triply charged precursors, and the ”ion types” to fit your experiment, typically y or y,b. Select the product ions for Skyline to display. We have chosen m/z > precursor, 3 ions, and N-terminal to Proline as shown in Figure 3.
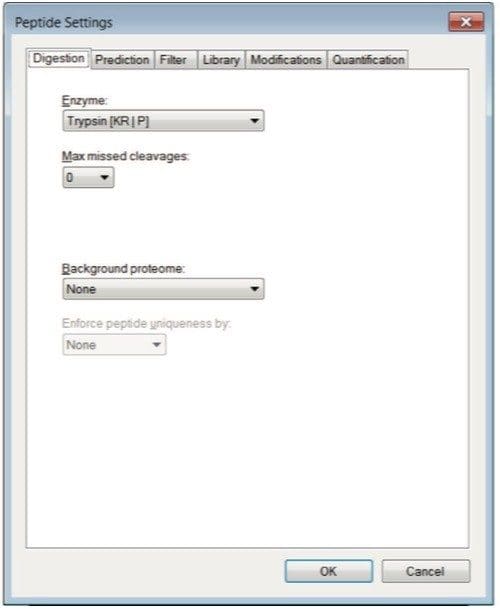
Step 2. When performing in silico digestion, the parameters should be set to match your experiment. For example, under Settings > Peptide Settings > digestion tab the default choice is Trypsin as the enzyme and zero Max missed cleavages (Figure 4).
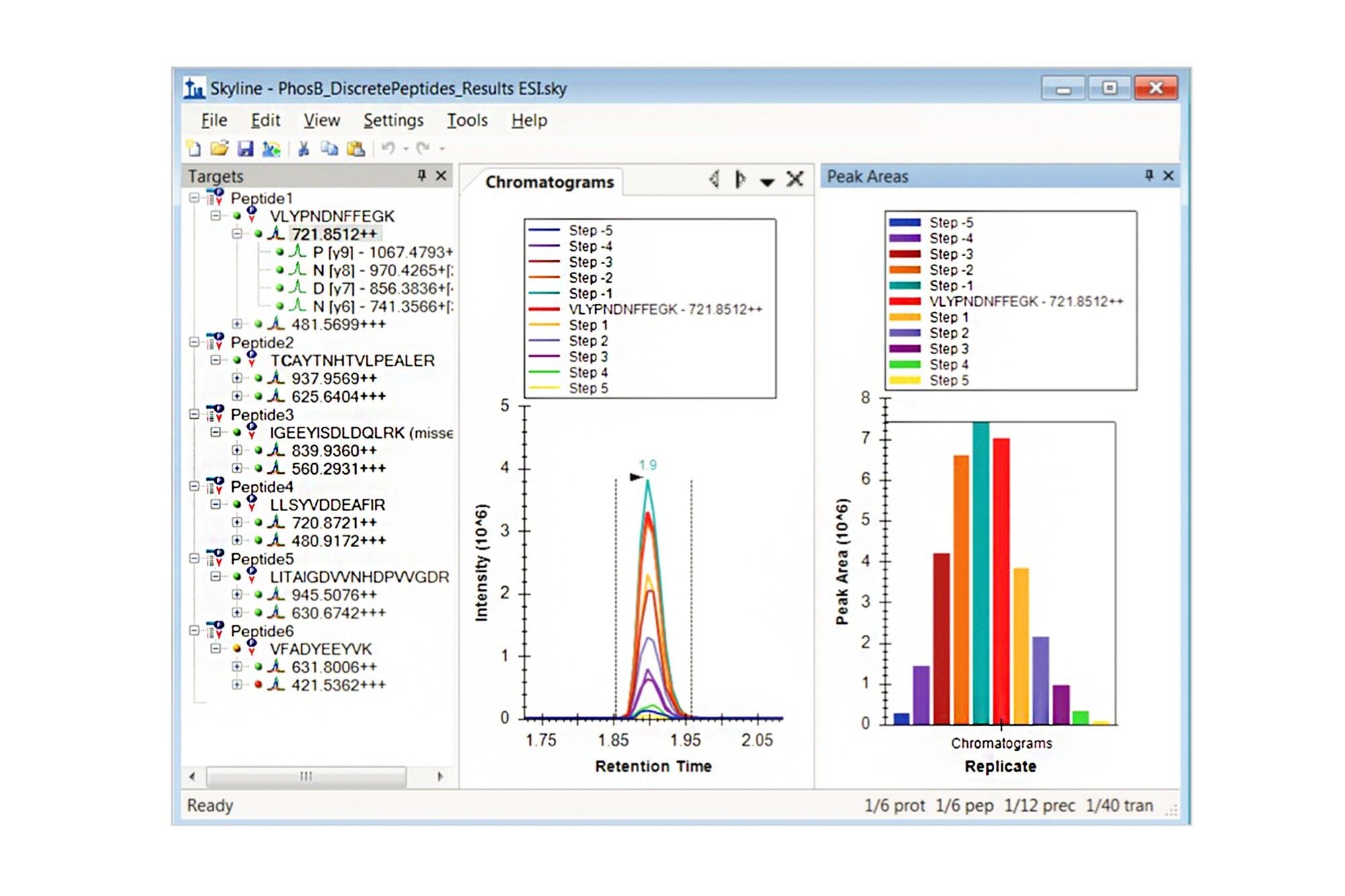
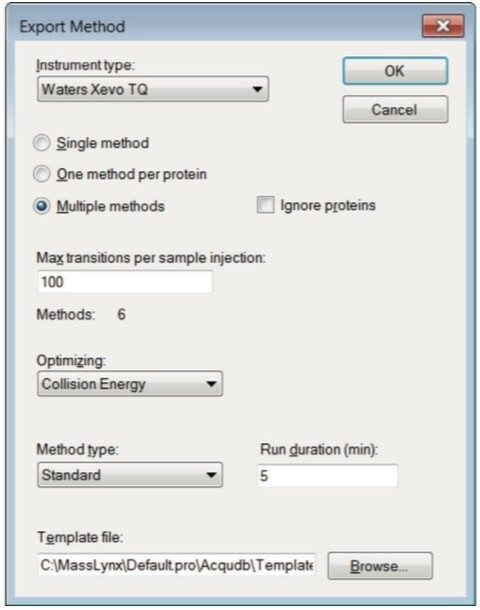
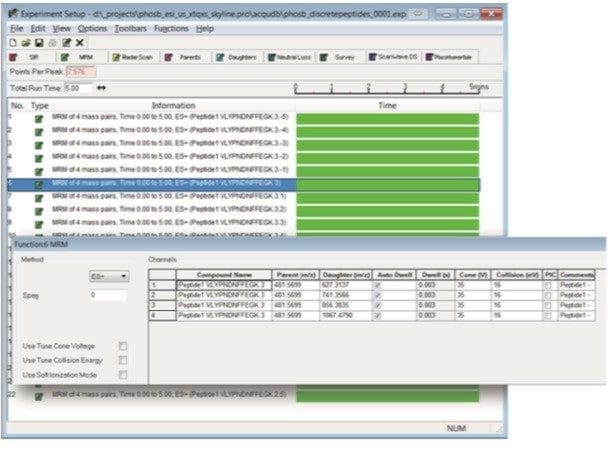
Step 3. Populate the peptide list by copying and pasting peptides individually in a list, or by importing a FASTA file. If you are working with proteins, you may paste the protein sequence to allow Skyline to perform in silico digestion. Create the MRM methods for injection under MassLynx by selecting File > Export > Method. Choose Waters Xevo for instrument type, select “Multiple methods”, “Max transitions per sample injection” = 100, select “Collision Energy” for “Optimizing,” “Method type” is “Standard”, and browse in an MRM method template (Figure 5). With these settings, our optimization will be complete in 6 injections taking a total of 30 minutes. Figure 6 shows an example of the MS method created in Skyline. In this method there are 22 functions with a total of 88 transitions to be monitored.
Additional notes: “Max transitions per sample injection” is a parameter that can be adjusted according to the quality and concentration of sample as well as length and resolving power of the LC method. Choosing too high of a number of transitions can impact the data quality by reducing the number of points per peak in the optimization. Additionally, the step size and step count can also be modified to ensure there are enough points per peak in the acquisition method created by Skyline. A consideration for shortening the number of injections is to use Scheduled chromatography in the method type. Scheduling will create time windows for the MRM transitions around the predicted retention time of a peptide and therefore more transitions can be added into a single MS method without compromising data quality.
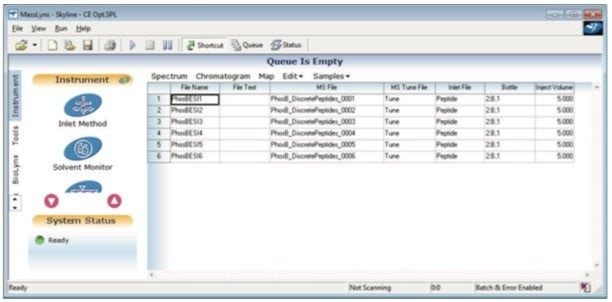
Step 4. Create the Sample List in MassLynx, browse in the individual MS methods and perform the injections (Figure 7).