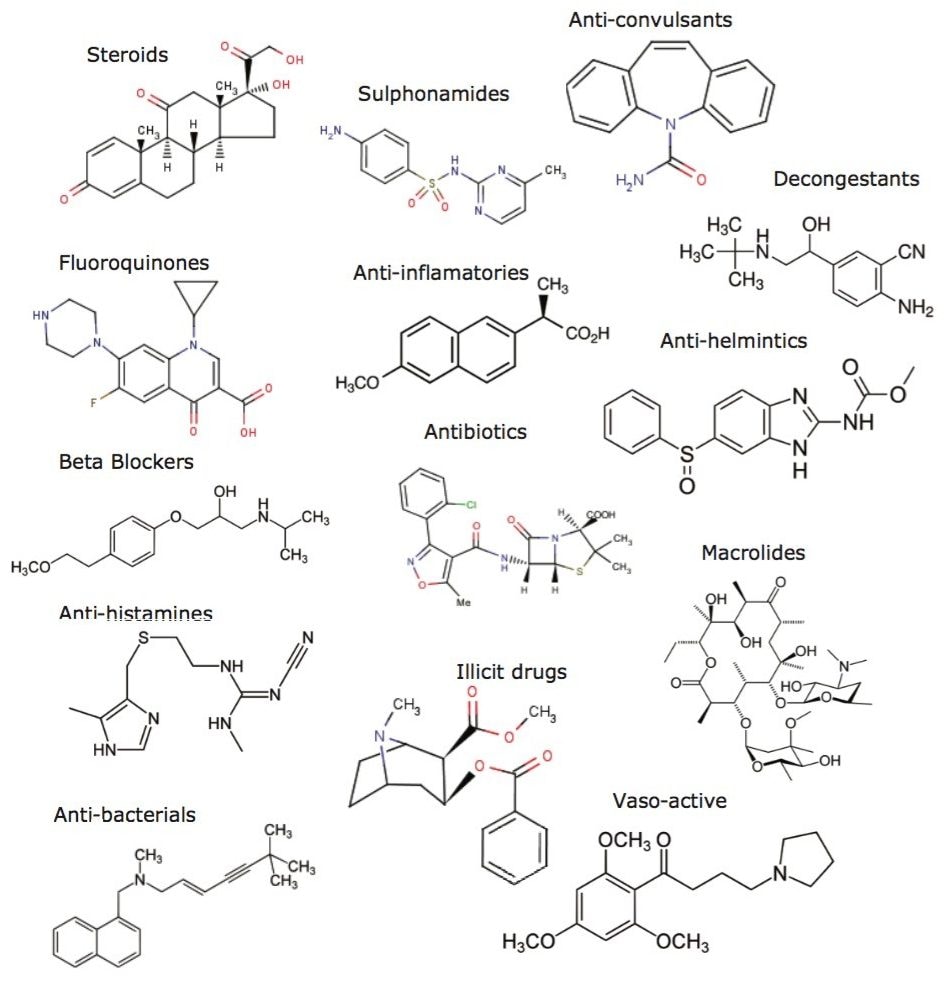
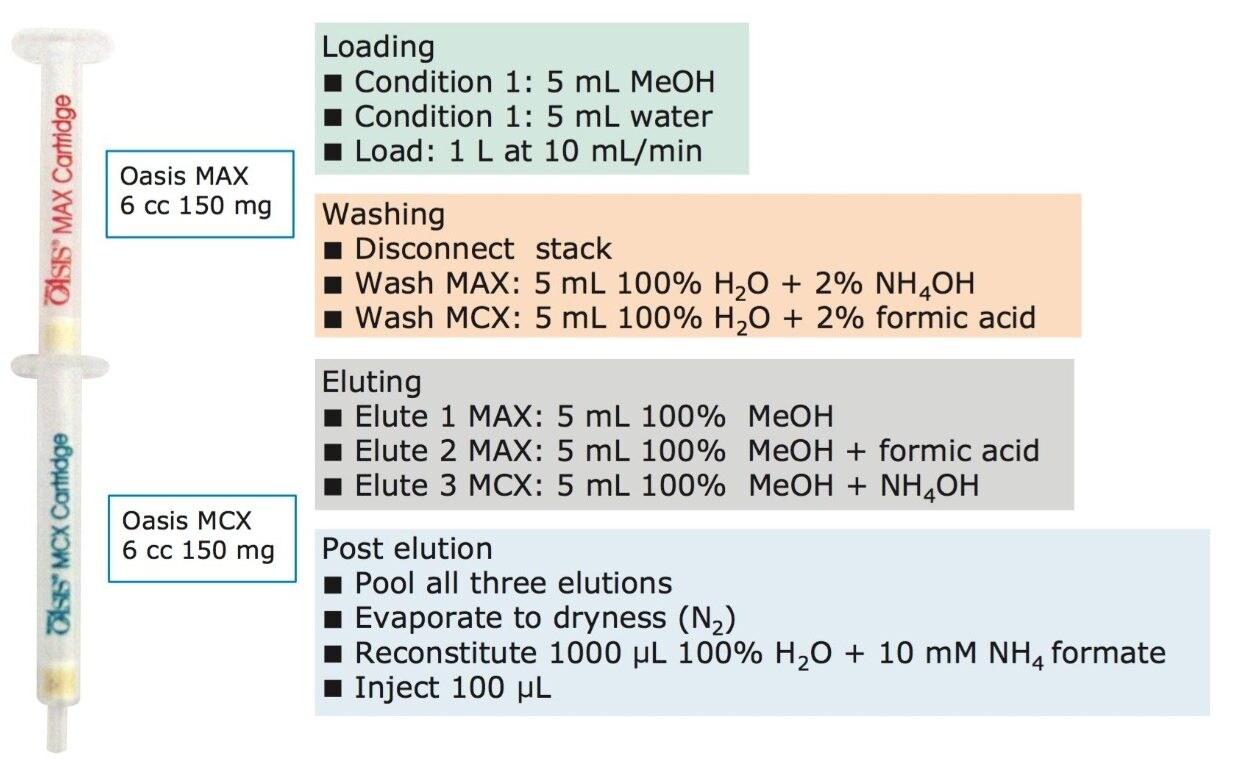
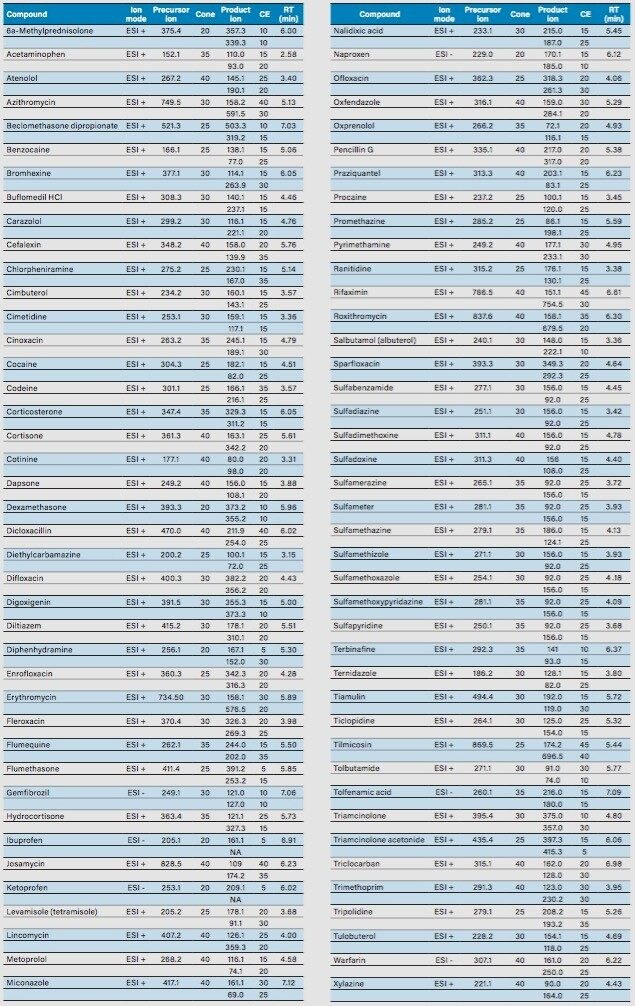
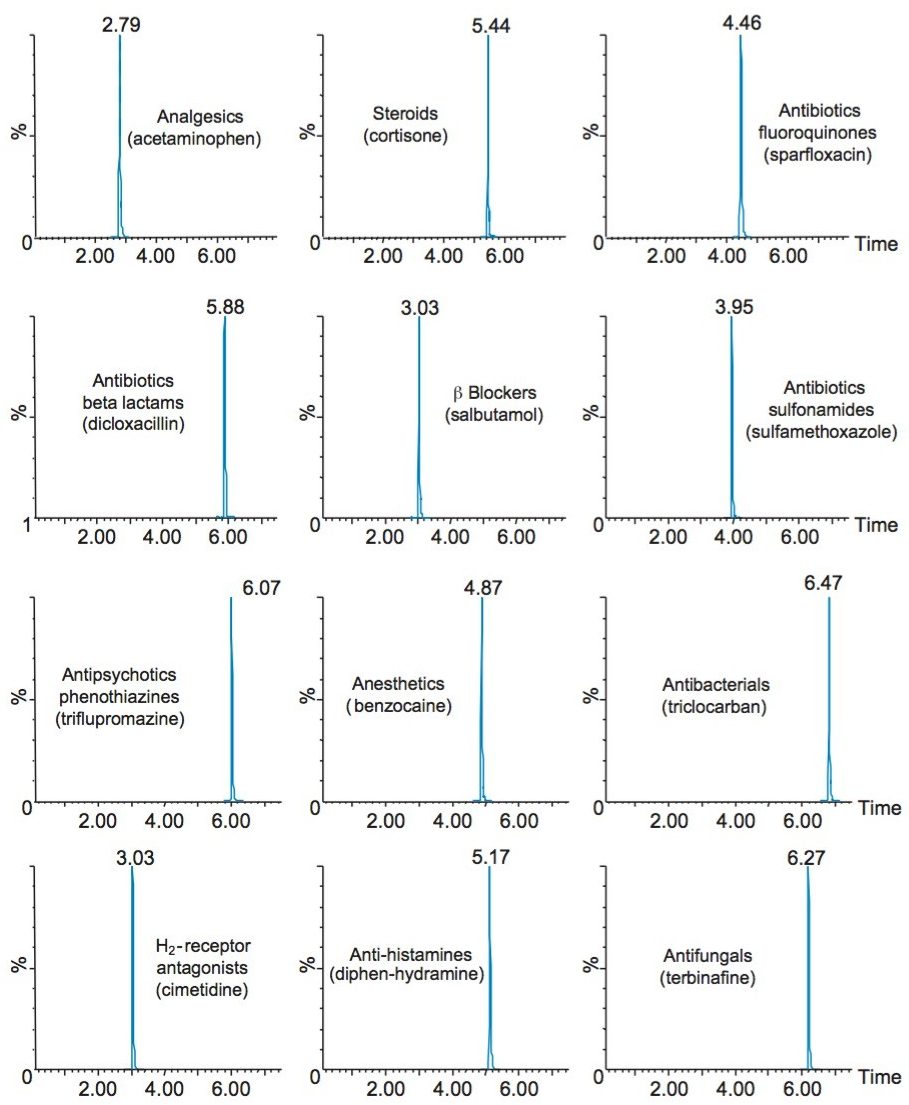
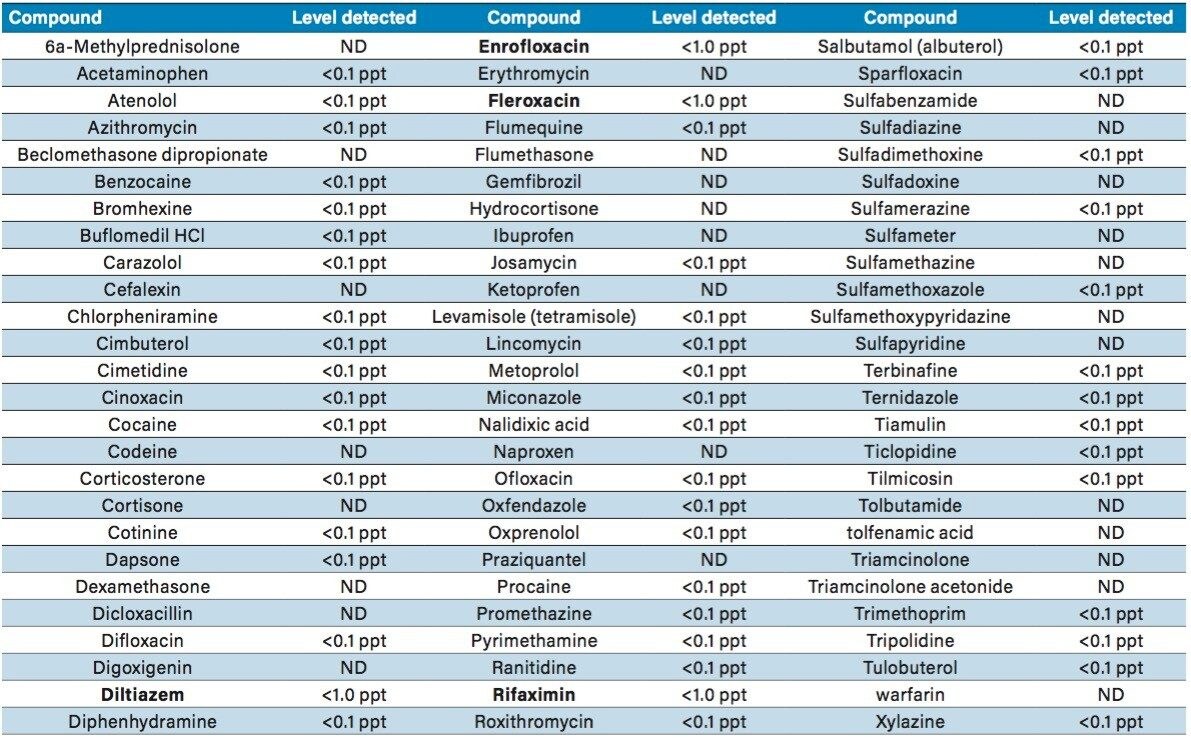
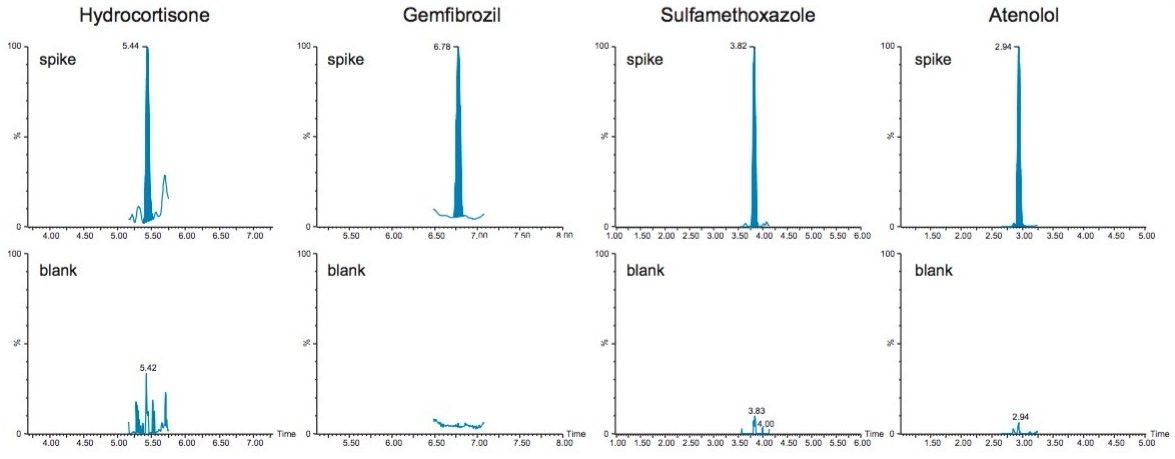
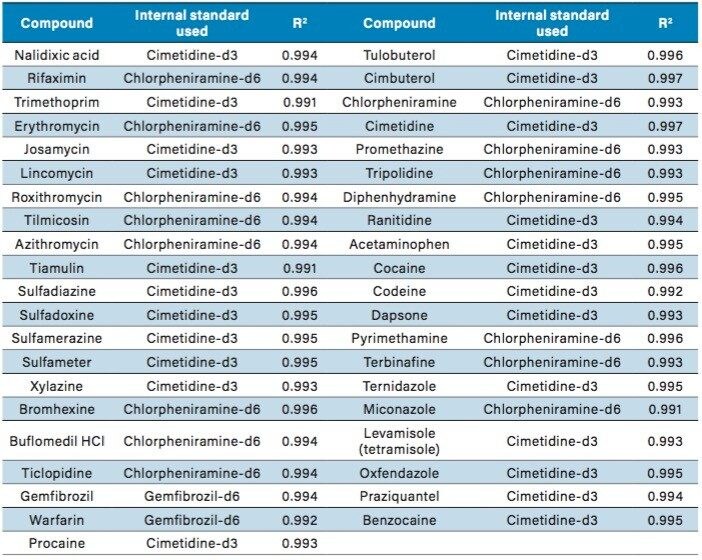
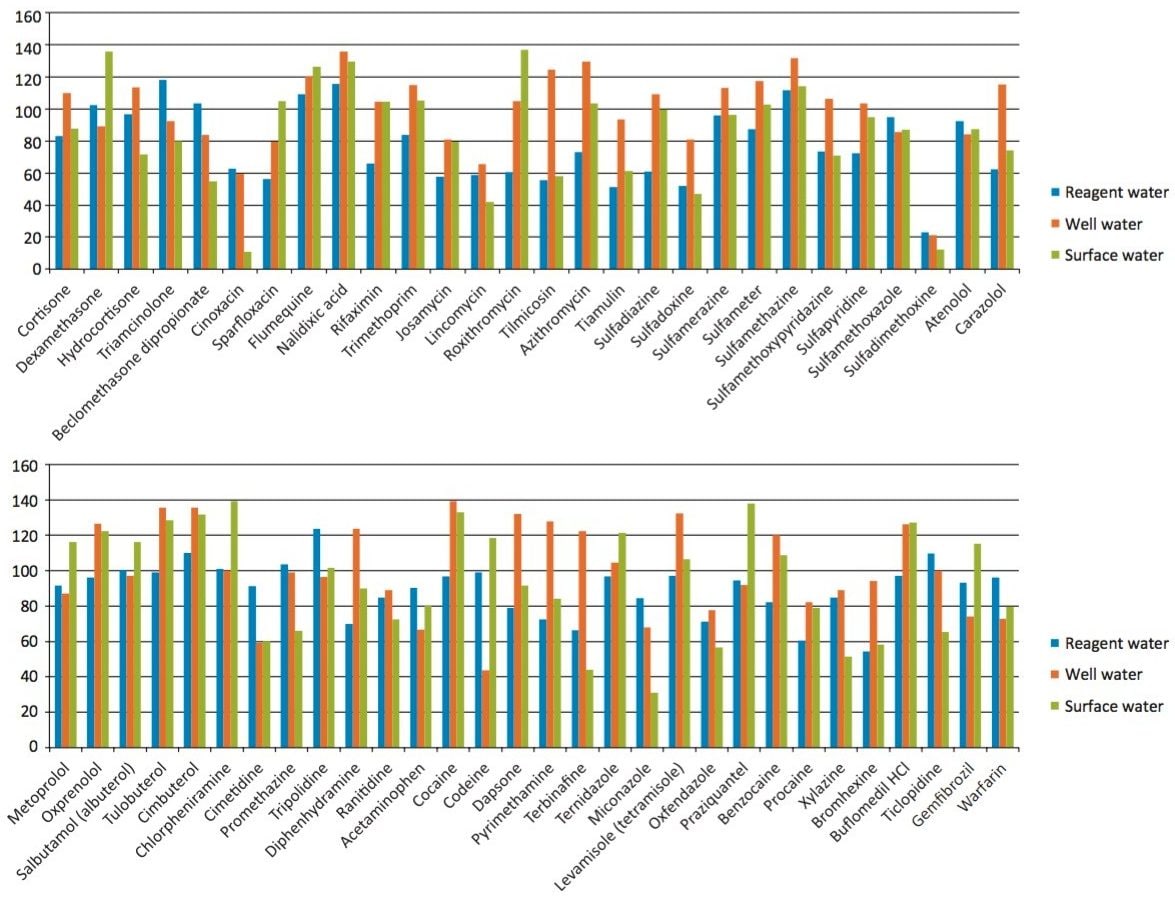
Sample preparation
The extraction process was performed using a tandem cartridge configuration with a Waters 6-cc Oasis MAX (p/n 186000369) and a 6-cc Oasis MCX (p/n 186000256) SPE Cartridge. This configuration allows for a three-tiered extraction mechanism that uses reversed-phase, anion exchange, and cation exchange. The extraction protocol was designed to ensure retention of acidic, basic, and neutral PPCPs. The Oasis MCX Cartridge was connected below the Oasis MAX Cartridge, and both were conditioned by passing through 5 mL of methanol followed by 5 mL of water. The water samples (1 L) were loaded at 10 mL/min onto the dual stack by vacuum using a bottle-to-SPE adapter. Once the loading step was completed, the cartridge stack was disassembled and each cartridge followed specific wash and elution steps, as shown schematically in Figure 2. The Oasis MAX Cartridge was washed with 5 mL of 5% ammonium hydroxide in water. The elution was performed in two steps:first with 5 mL of methanol (neutral PPCPs), and second with 5 mL of methanol containing 5% formic acid (acidic PPCPs). Both elution fractions were collected in a 20-mL glass tube. The Oasis MCX Cartridge was washed with 5% formic acid and eluted with 5 mL methanol containing 5% ammonium hydroxide (basic PPCPs). The MCX and MAX elution fractions were pooled and evaporated to dryness at 60 °C under a gentle stream of nitrogen. The dried eluate was reconstituted with 900 μL (2 x 450 μL) 10 mM ammonium formate. The internal standard mix (100 μL) was then added to give a final concentration of 1.0 ppb. Matrix-matched calibration standards were prepared with the same protocol with the exception of the final eluate, which was reconstituted in 800 μL (2 x 400 μL) 10 mM ammonium formate, and 100 μL of the internal standard mix was added. The final 100 μL was utilized to post spike 100 μL of the PPCP mix at various concentrations in 10 mM ammonium formate. The standards for the majority of compounds were spiked at concentrations ranging from 0.1 to 5.0 ppb (0.1, 0.2, 0.25, 0.5, 1.0, 2.0, 2.5, and 5.0 ppb final concentration). This range equates to 0.1 to 5.0 ppt in the original sample. 13 compounds demonstrated higher limits of detection and were therefore analyzed from 1.0 to 50.0 ppb (equivalent to 1.0 to 50.0 ppt in the water samples). These compounds were cefalexin, cinoxacin, codeine, corticosterone, dicloxacillin, erythromycin, gemfibrozil, ibuprofen, ketoprofen, naproxen, tolfenamic acid, triamcinolone, and warfarin. The internal standard mix consisted of three isotopically labeled standards:
Cimetidine-d3-N-methyl-d3, Chlorpheniramine-d6-maleate-N,N dimethyl-d6, and Gemfibrozil-d6-2,2 dimethyl-d6 (purchased from C/D/N Isotopes Inc.).