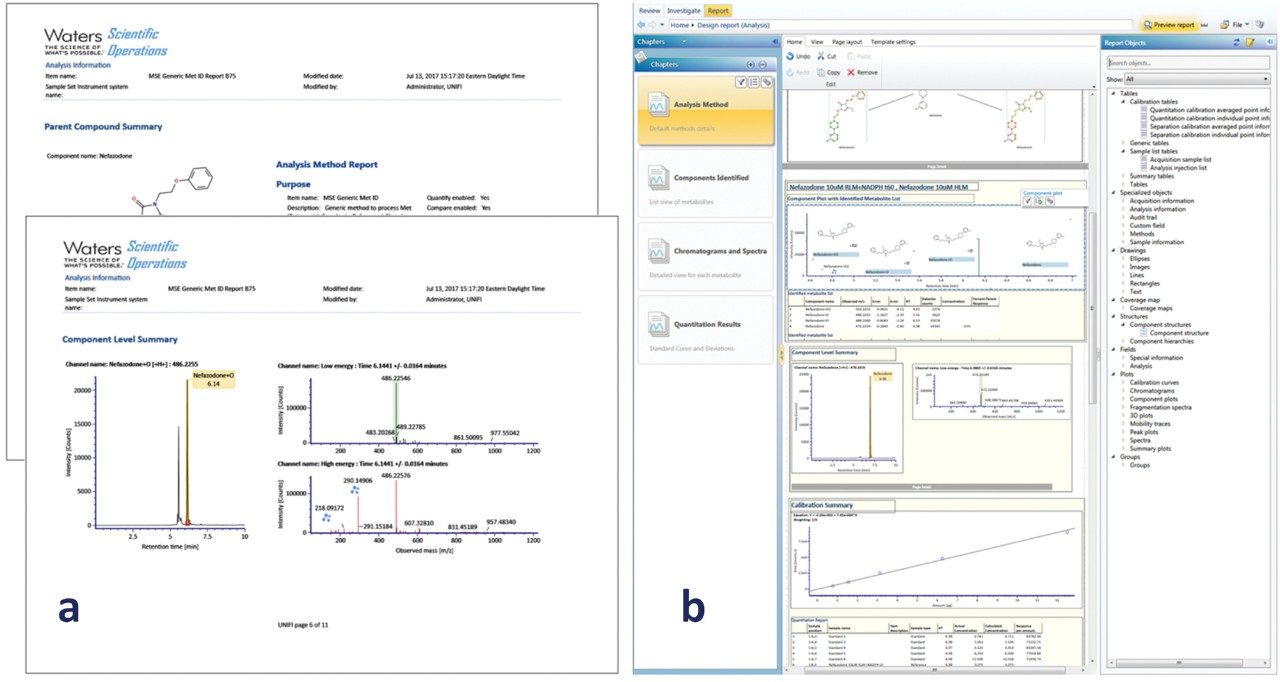
Metabolite identification has evolved in recent years from a largely labor intensive manual process to a technique that leverages software, which can rapidly screen and detect metabolites from in vitro or in vivo studies. Screening also requires close integration with tools that enable accurate quantification, trend plotting, and reporting. Projects that involve challenging drug concentrations, complex pathways, drug delivery systems, or biotherapeutic molecules may also require additional analytical tools such as ion mobility, targeted modes, and/or tailored workflows.
The analysis of data generated from ion mobility-equipped instrumentation has historically required separate software packages and processing workflows. UNIFI and Vion IMS QTof implement this in a straightforward, integrated manner, enabling easy access to cleaner data with high quality, excellent mass accuracy, and robust CCS measurements.
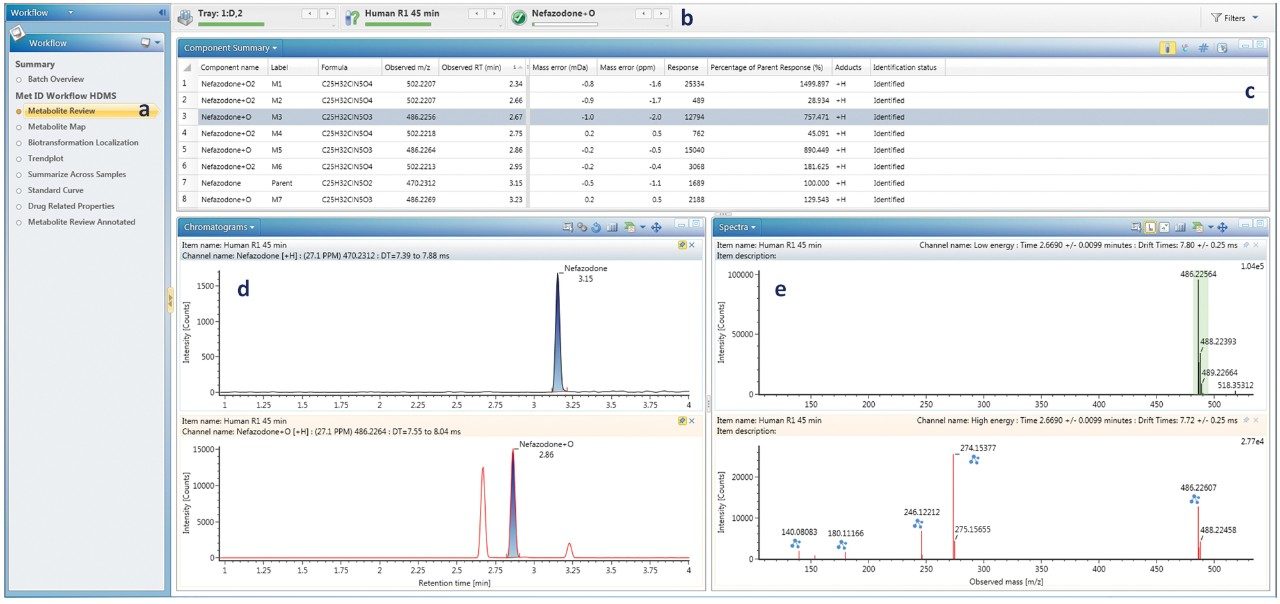
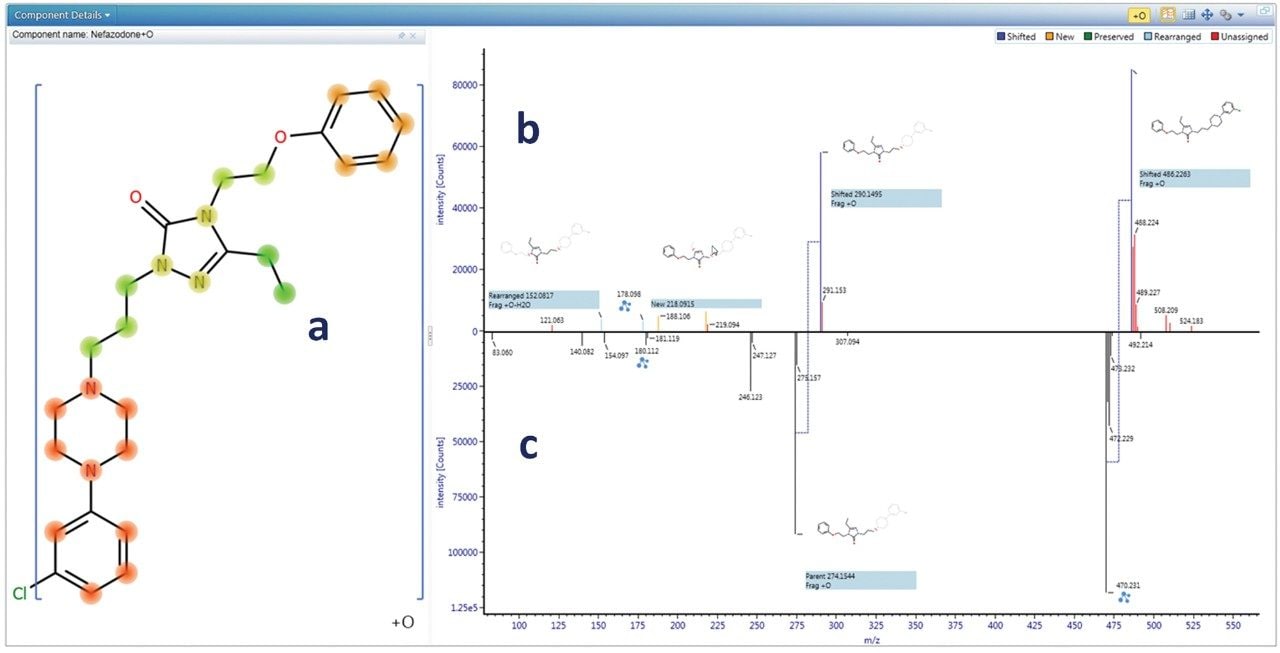
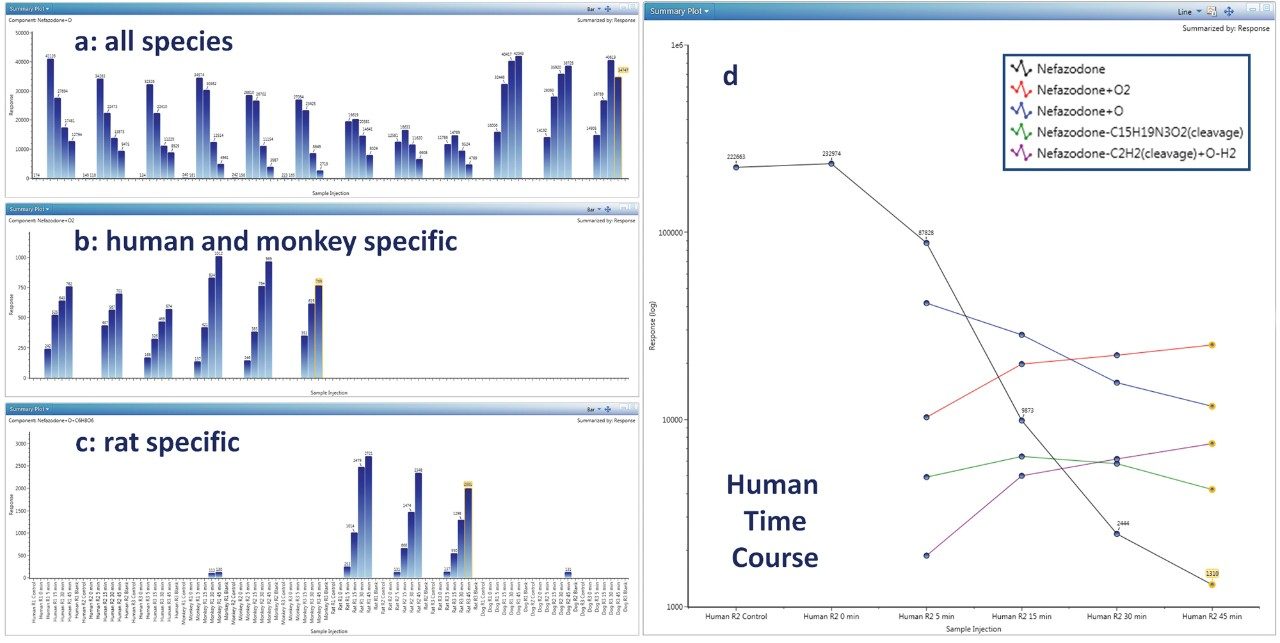
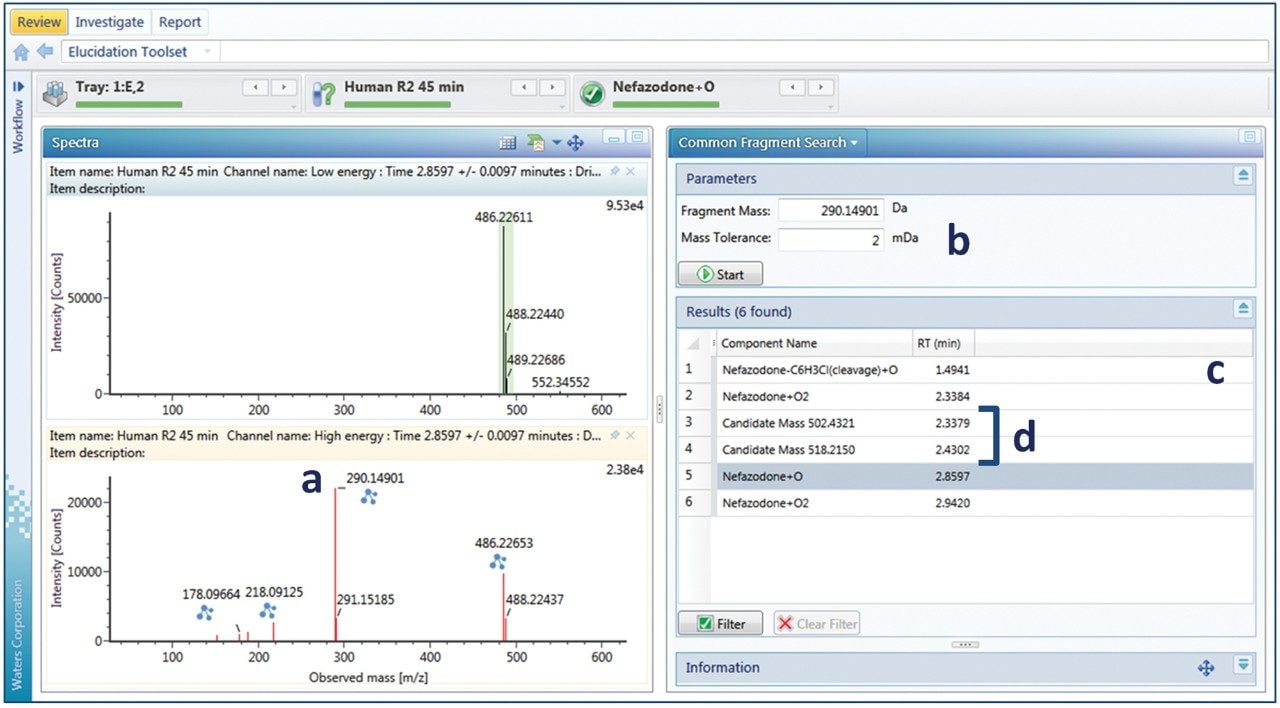
This application note outlines the use of integrated quantitative and qualitative processing for managing multispecies studies. UNIFI enables advanced features such as: flexible viewing of samples/species/time data; trend plot tools for visualizing studies; use of scientific libraries to store and retrieve identified compounds; and comprehensive tools for analyzing unknowns and characterizing metabolites and impurities.