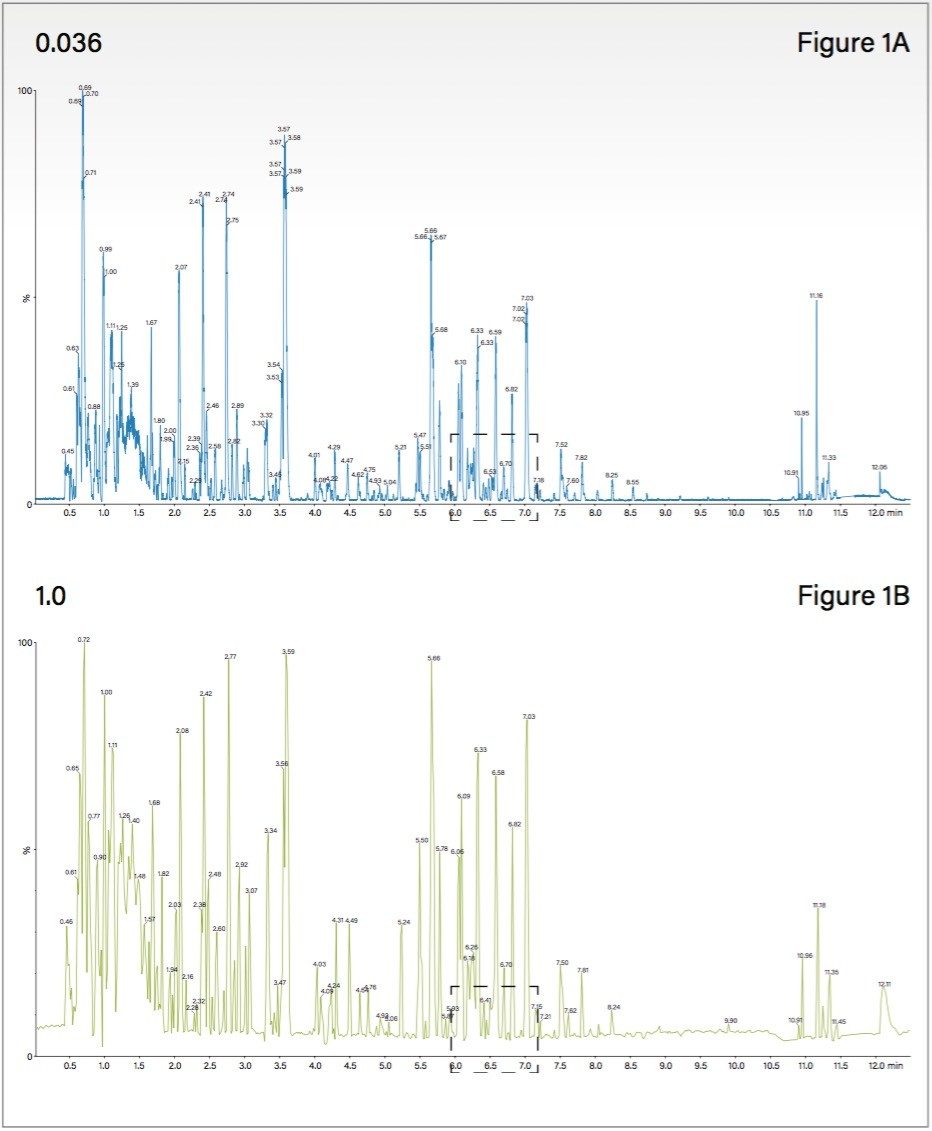
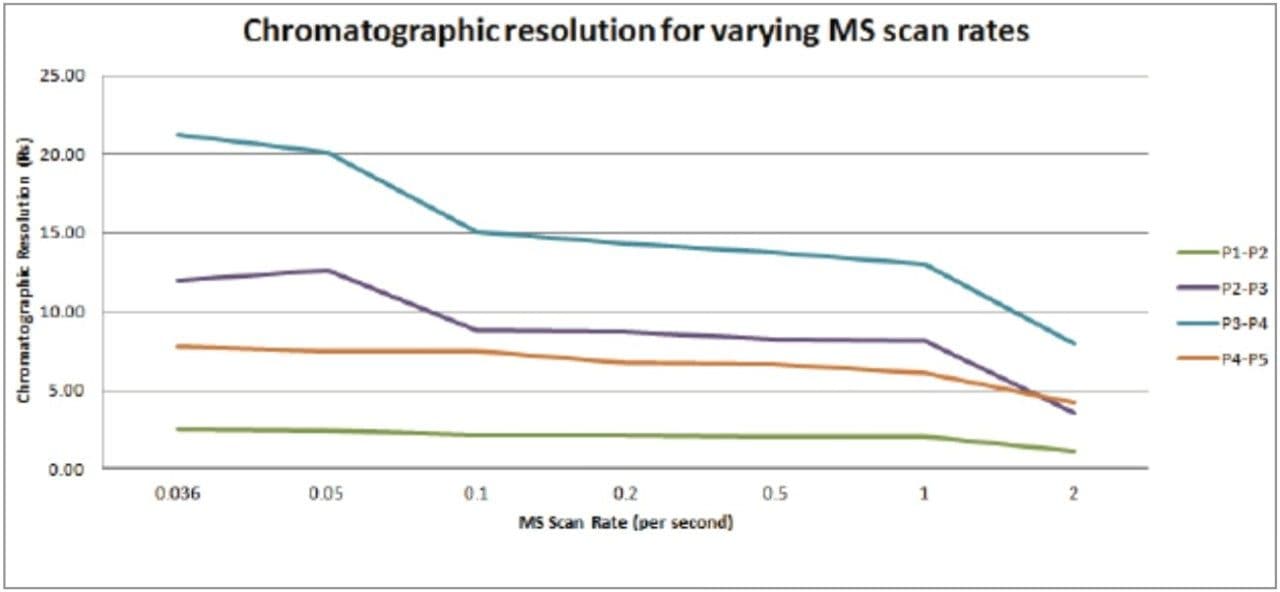
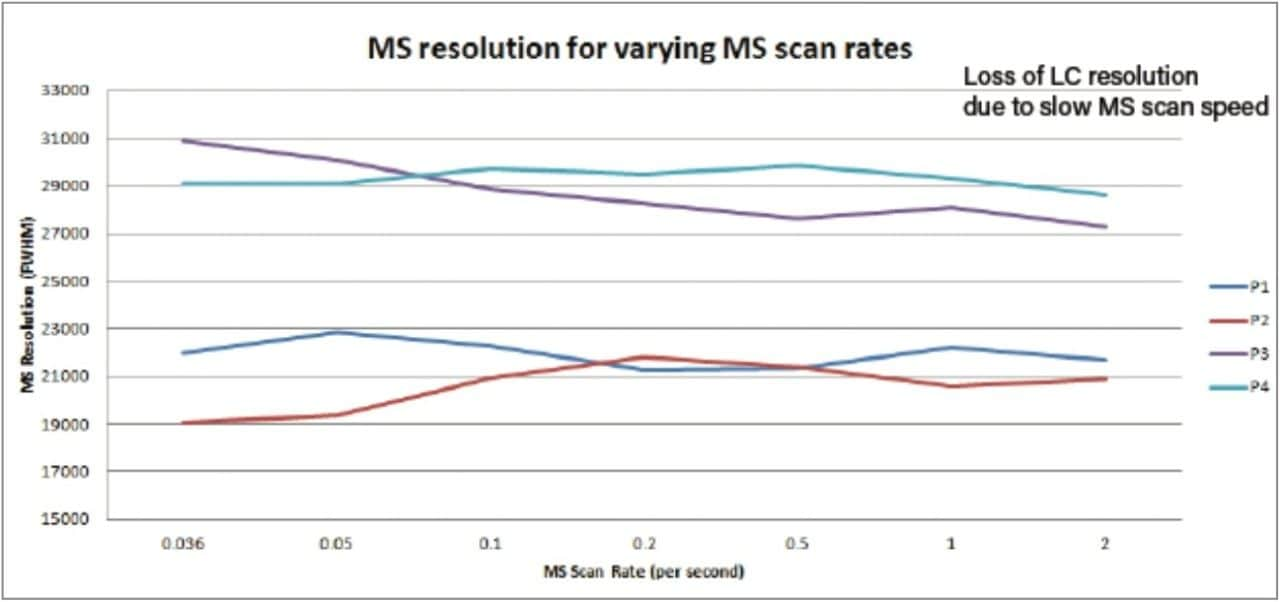
The MS data acquisition rate or scan time selected is usually determined by the design of the mass spectrometer and the MS resolution required for the analysis.
Fourier-transform mass spectrometers (FTMS) such as Fourier-transform ion cyclotron resonance mass spectrometry (FTICR) and Orbitraps have the potential to reach extremely high MS resolution, greater than 200,000 full width at half maximum (FWHM). In order to achieve such values, they require acquisition times in excess of 1 second, and therefore these high-resolution MS spectral results are most beneficial for structural elucidation experiments. Such slow data acquisition times can be acceptable when the liquid chromatography (LC) analysis time is greater than 45 minutes and when LC peak widths are greater than 10 seconds at the base. As the data acquisition time is reduced (faster acquisition) on a FTMS, there is commensurate reduction in MS resolution; this was demonstrated by Olah et al.1 where the use of MS data acquisition rate of 100 milliseconds resulted in an effective mass resolution of 10,000 FWHM.
The MS data acquisition rate can have a significant effect on the quality of the data obtained in complex mixture analysis experiments such as metabonomics, metabolomics and metabolic phenotyping. Metabolic phenotyping studies require the analysis of biological samples from a large cohort ranging from several hundred to several thousand samples. Therefore in these studies, both short run times and high chromatographic resolution are required. When operated in gradient mode, modern sub-2-µm particle LC systems produce chromatographic peaks with widths of 1–3 seconds at the base.2,3 It is therefore necessary to have a data acquisition rate that allows for accurate analyte quantification and identification when peaks are eluting rapidly and/or overlapping. For reliable analyte quantification, it is necessary to have 8–10 data-points across the chromatographic peak; therefore for a peak width of 1–3 seconds, a data acquisition rate of 50–100 milliseconds is required. Unlike FTMS instruments, Time-of-flight (Tof) mass spectrometers acquire MS data at virtually the same resolution independent of the data acquisition or scan rate.