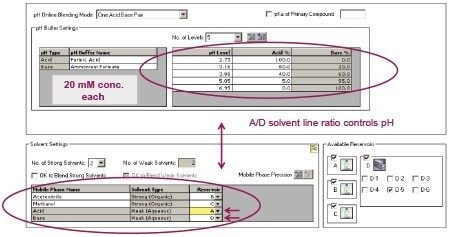
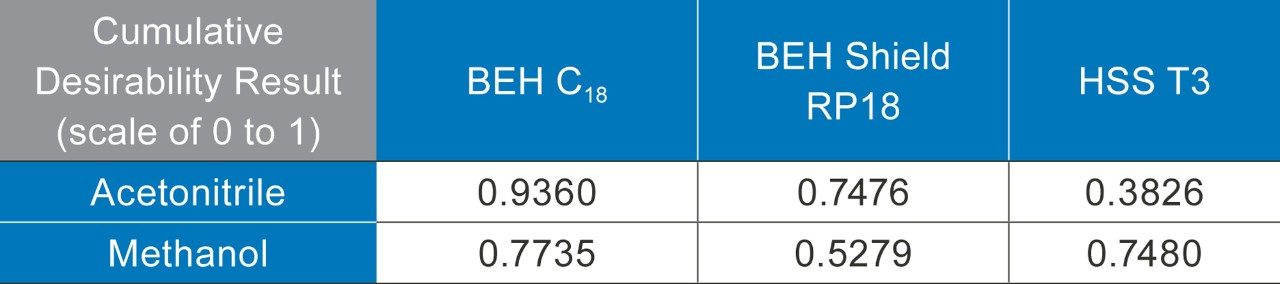
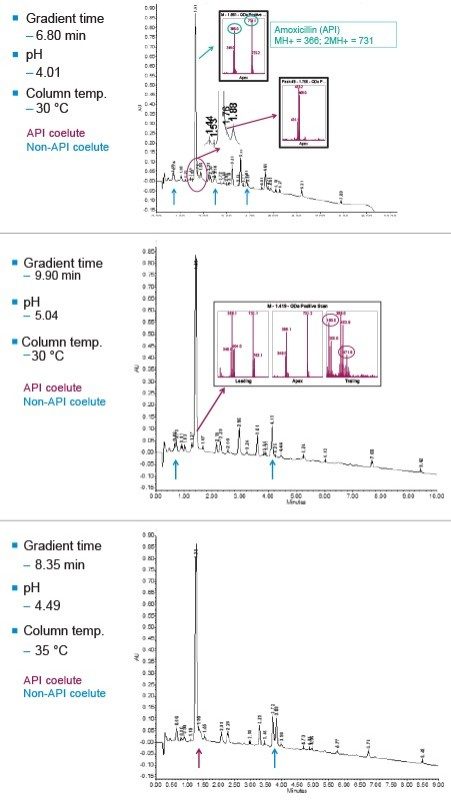
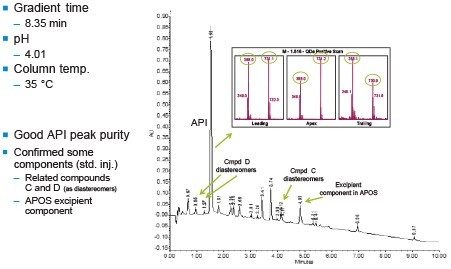
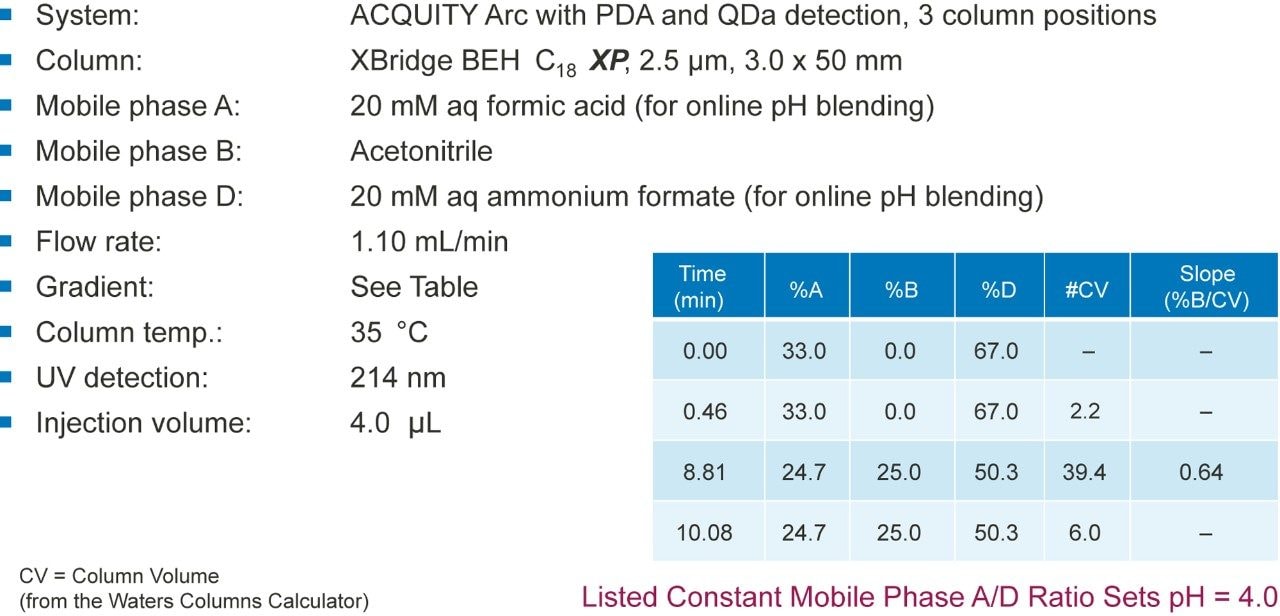
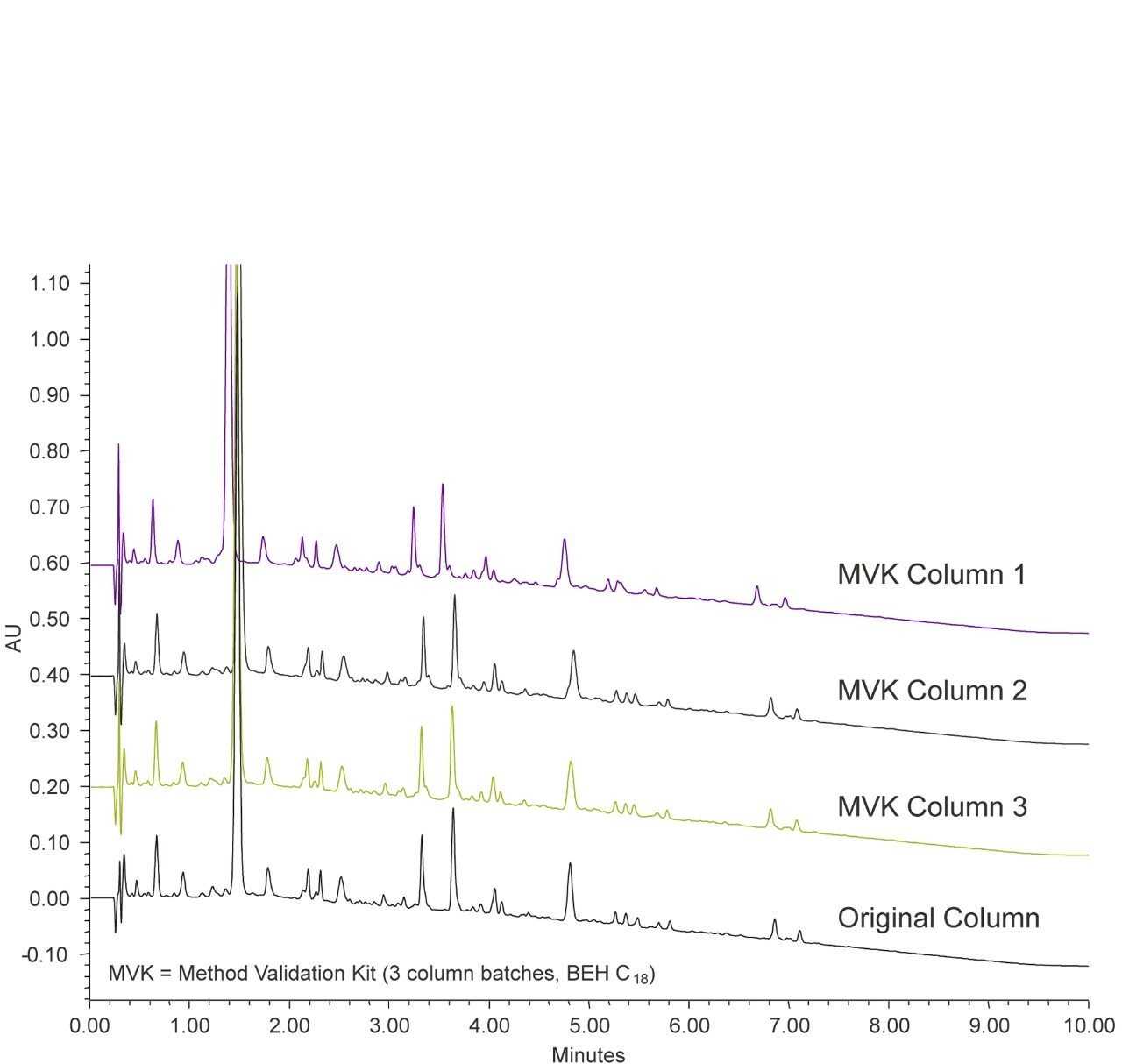
The development of an analytical chromatographic method has three discreet stages: Set up, screen, and optimize. In the set up stage, the analyst selects the factors to vary, the responses to measure, the response criteria to assess progress, and the strategy to follow. Factors are the “inputs” to method development that cause analyte selectivity and resolution changes. Some factors, like the strong solvent, pH, and column stationary phase, have stronger effects whereas others, like gradient slope3 and column temperature, have weaker effects. Furthermore, some factors are numeric and continuously variable, such as column temperature, pH, and gradient slope. Other factors are “categorical” or non-numeric, like column stationary phase. For each factor selected, the factor values are then set, such as which exact columns, solvents, and pH values to use.
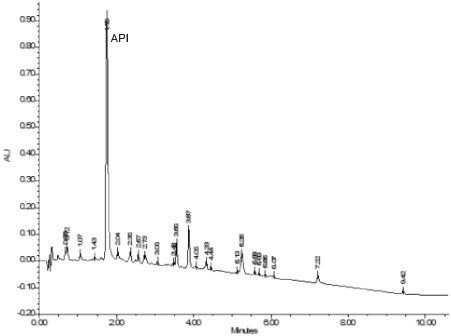
Responses are the “outputs” to method development, as measured from chromatograms. Some responses are specific peak properties such as retention time, resolution, and tailing for specific components. Other responses are chromatogram properties from peak counting (aggregation), as in the total number of peaks in a chromatogram or the number of peaks with a specific desirable result.
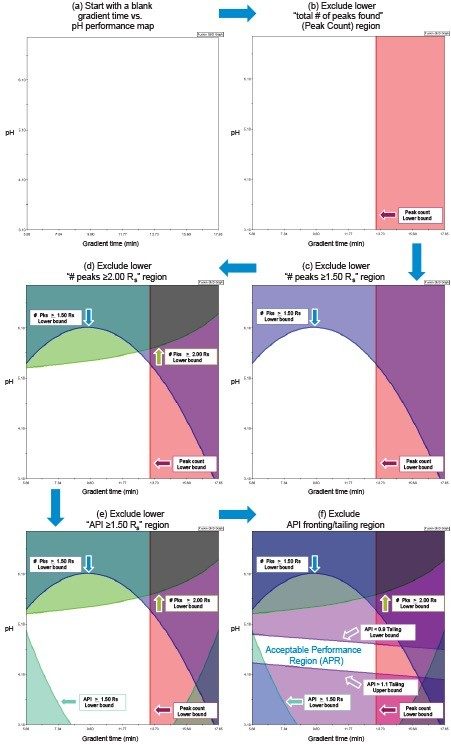
Response criteria are used to compare the quality of different factor combinations. Examples of chromatogram property response criteria include “total # of peaks in the chromatogram = # of components known or believed to be present” or “maximize # of peaks found”. For peak property response criteria, these include retention, tailing, and resolution in a specific range.
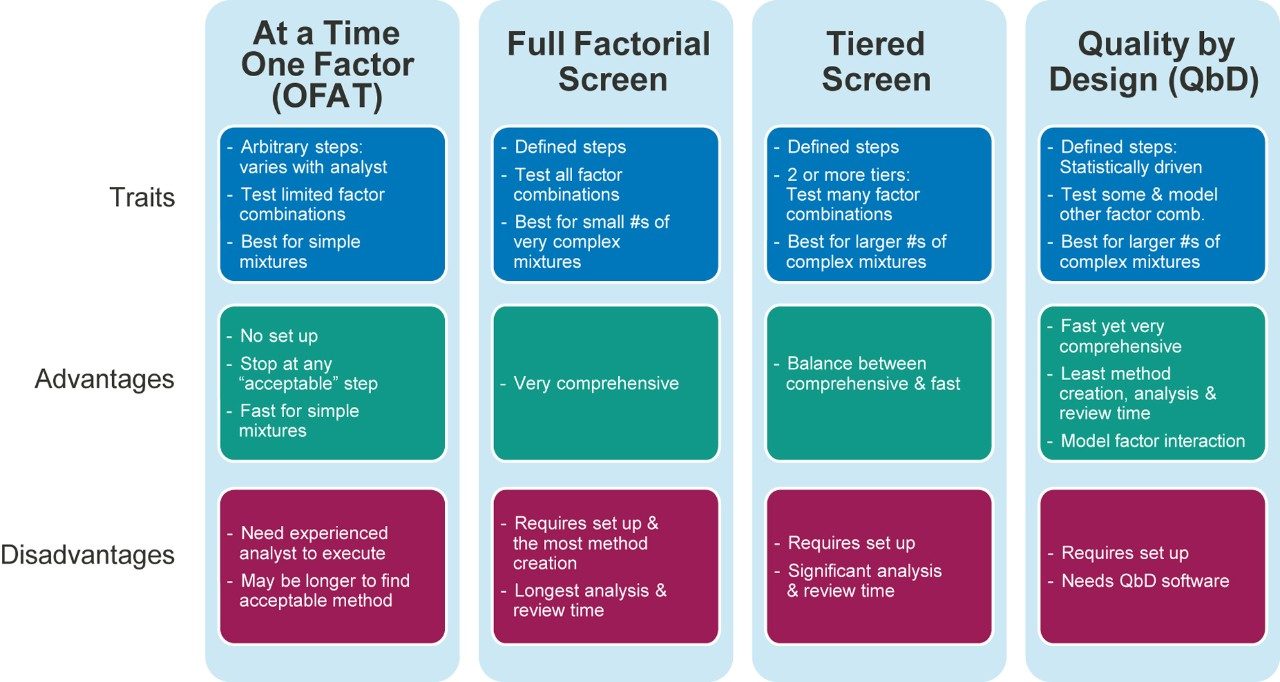
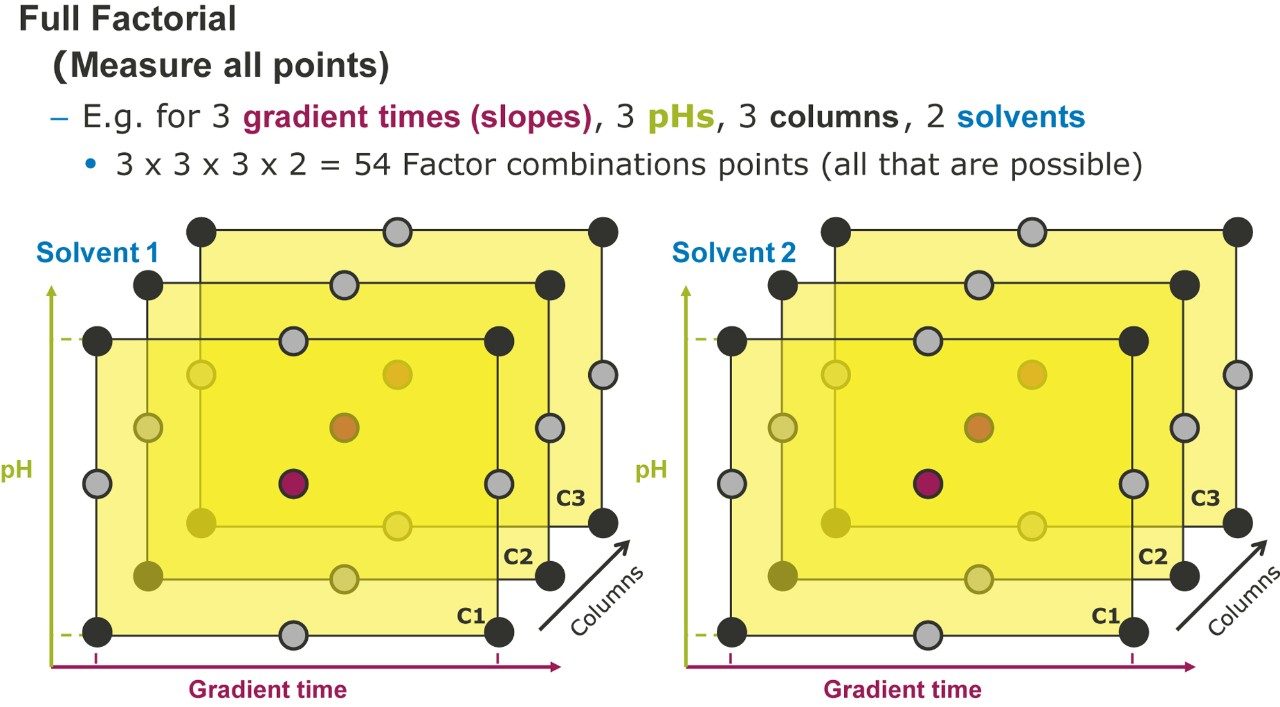
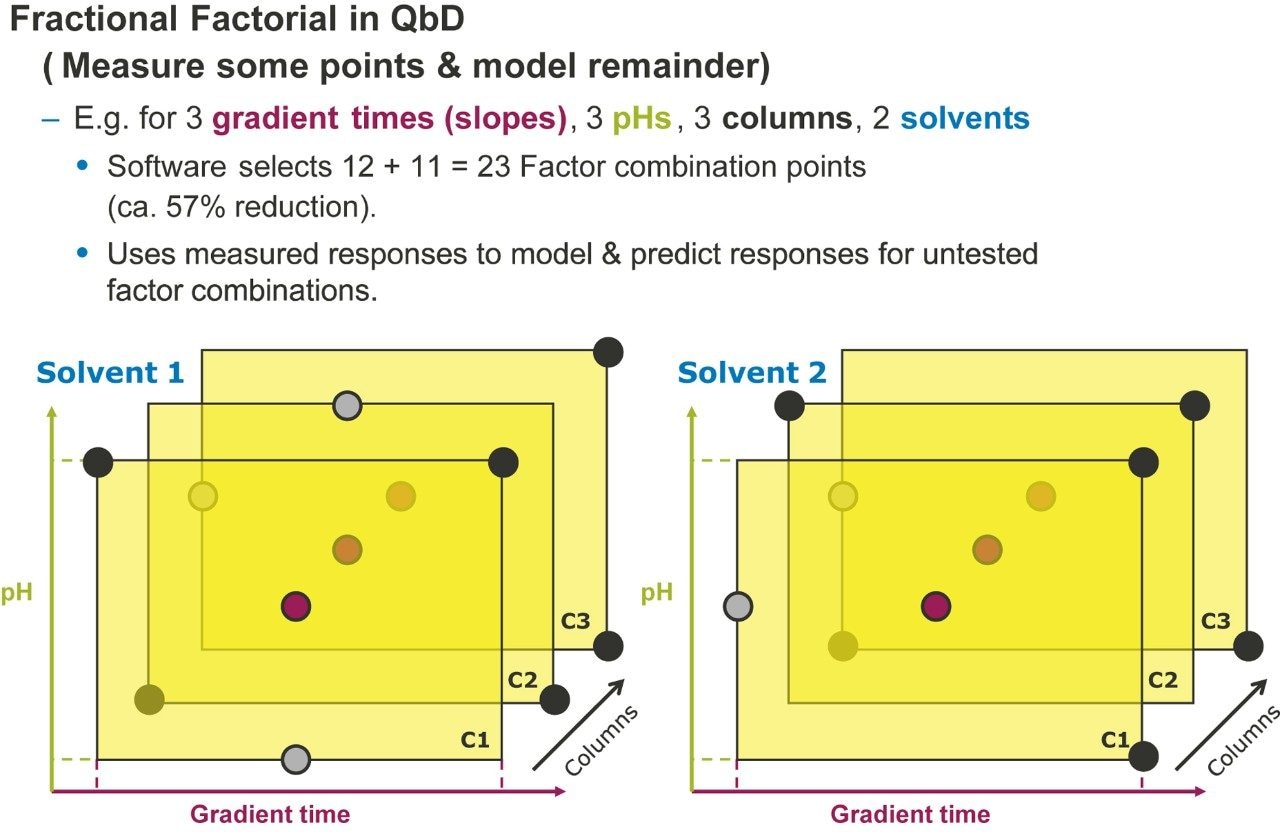
Figure 2 lists four method development strategies. The simplest strategy to execute but the least comprehensive is “One Factor At a Time” or OFAT, where the analyst alternates between picking one factor and value to test and assessing the responses from that test. The most comprehensive and labor/time intensive strategy uses a Full Factorial Screen where all combinations of factor values are tested to determine the best combination. A hybrid of these two approaches is the Tiered Screen. Here, all factors except one are locked to a specific value (e.g. a single specific column stationary phase, strong solvent, temperature, etc.). The one factor allowed to vary is changed to assess its affect on the responses. For example, the pH may be varied between a low and a high value. Based on how well response criteria are met, the one factor varied in the first tier is fixed to a single value. Moving to the second tier, some of the original locked factors are now systematically varied in full factorial fashion. This strategy is a balance between being comprehensive and being fast.