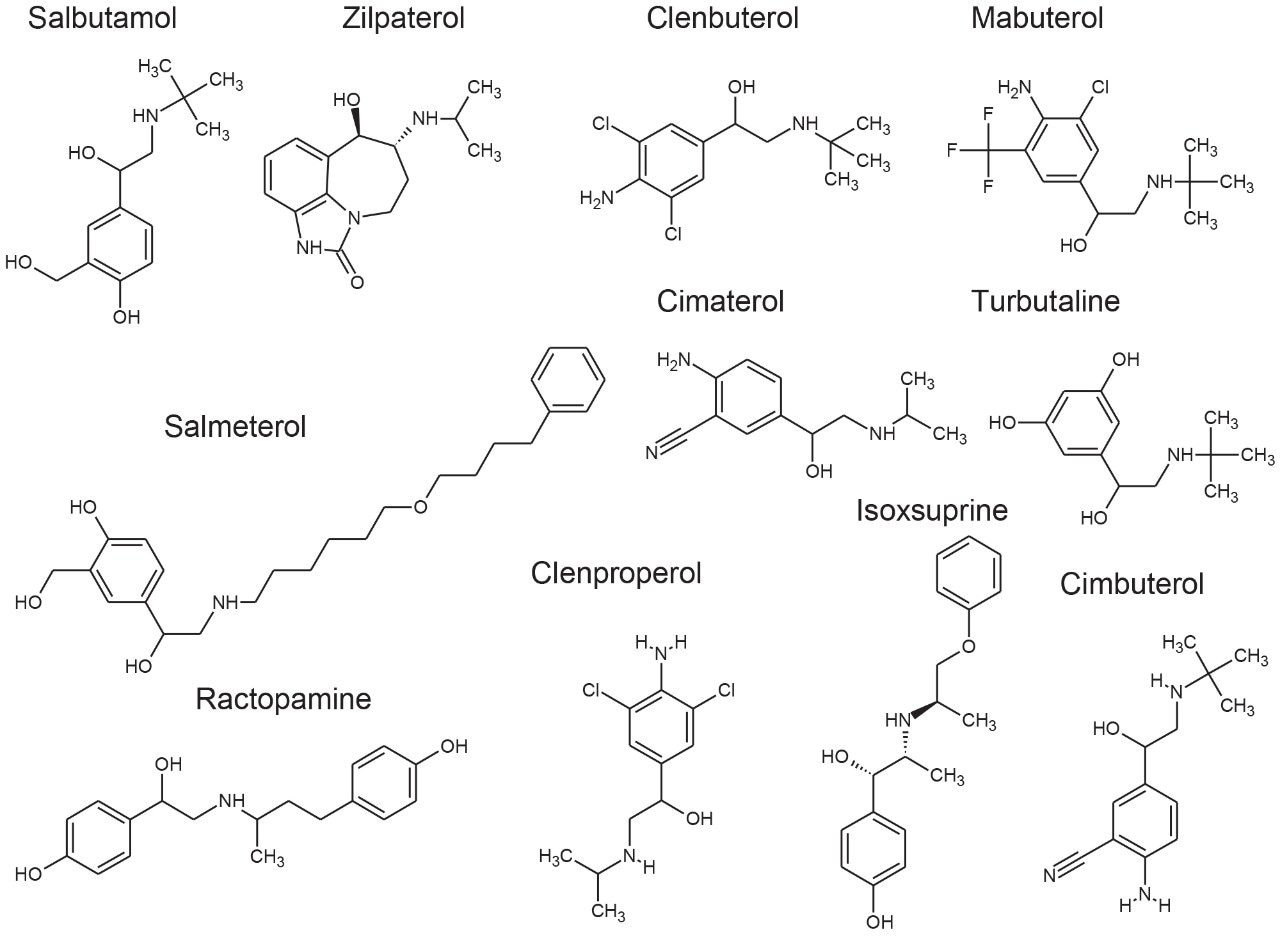
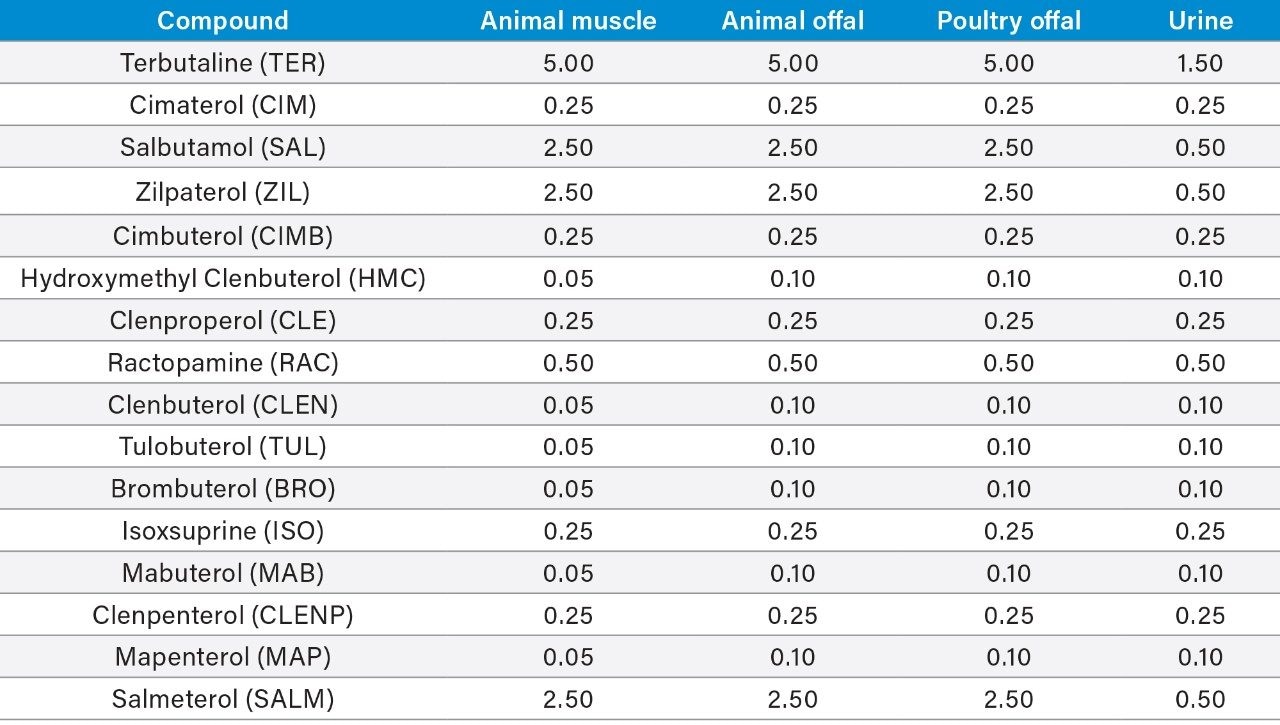
Although the administration of β-agonists as growth-promoting agents in food-producing animals is banned in many countries due to concerns over human health, there are exceptions.2 Ractopamine and zilpaterol are authorized for the production of some animals (e.g. cattle and pigs) in a limited number of countries such as the U.S., Canada, and Brazil. The Joint FAO/WHO Expert Committee on Food Additives (JECFA) has established Maximum Residue Limits (MRLs) for ractopamine in cattle and pig muscle (both 10 µg/kg), which have been adopted by the Codex Alimentarius Commission (Codex) and implemented in some countries (e.g. Canada).
Others have alternative limits for ractopamine, such as the tolerances set in the U.S. (30 and 50 µg/kg in cattle and pig muscle, respectively).
However, many countries disagree with the Codex standards and are restricting or banning meat products containing β-agonists. In the EU, MRLs only exist for clenbuterol (cattle and horse). Other β-agonists are prohibited for use in food producing animals. As no Minimum Required Performance Limits (MRPLs) were set for these banned β-agonists,3 the EU relies on Recommended Concentrations (RCs) to indicate required performance of associated analytical methods. In contrast to MRPLs, RCs have no legal standing and are used for guidance only.
Importing countries can set even lower limits for testing based upon trading decisions to provide better warranties to their customers and to gain commercial advantage. For example, Russia established an action level of 0.1 µg/kg for ractopamine in imported beef consignments; one hundred times lower than Codex MRL. However, as Russia has a zero tolerance policy, if a laboratory found any confirmed residues at lower concentrations, the consignment would be rejected. This drives down the analytical limits for those involved with pre-export testing.
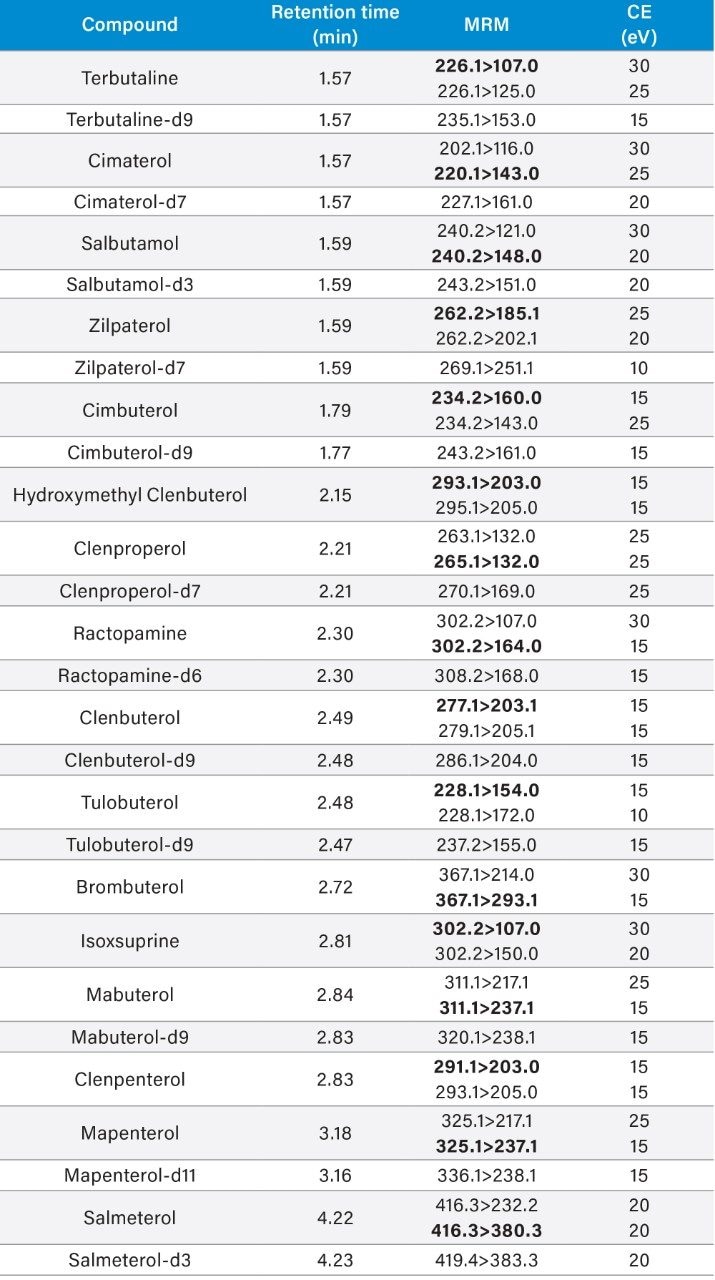
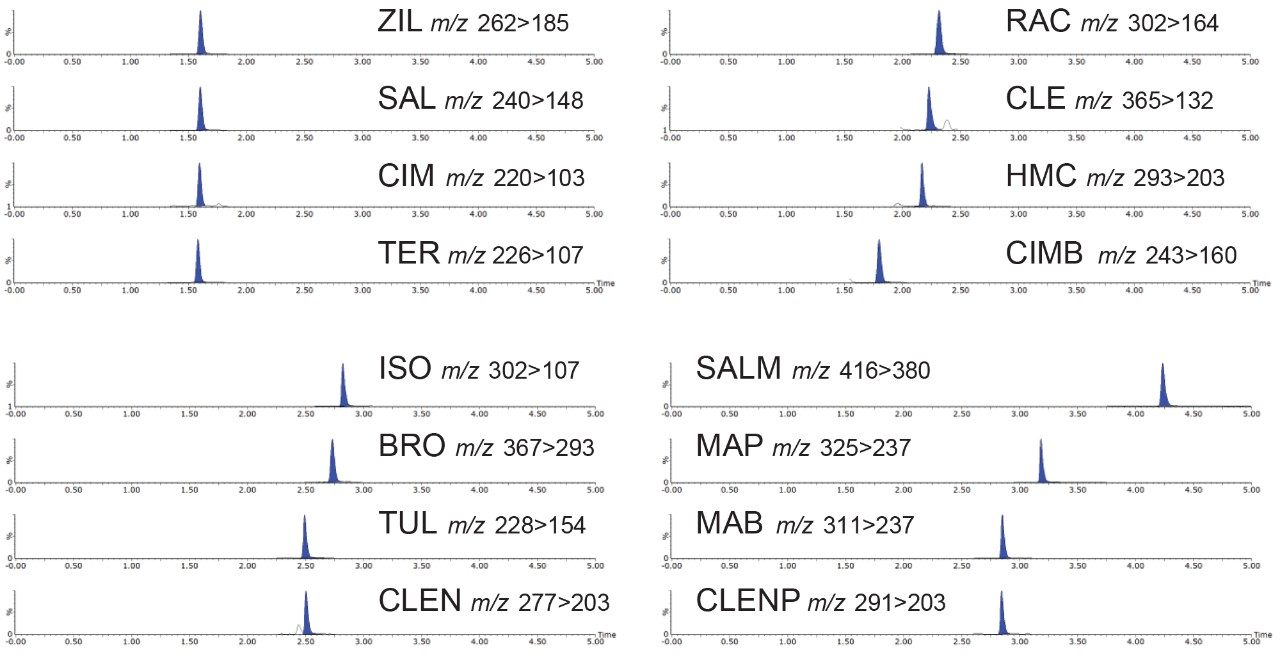
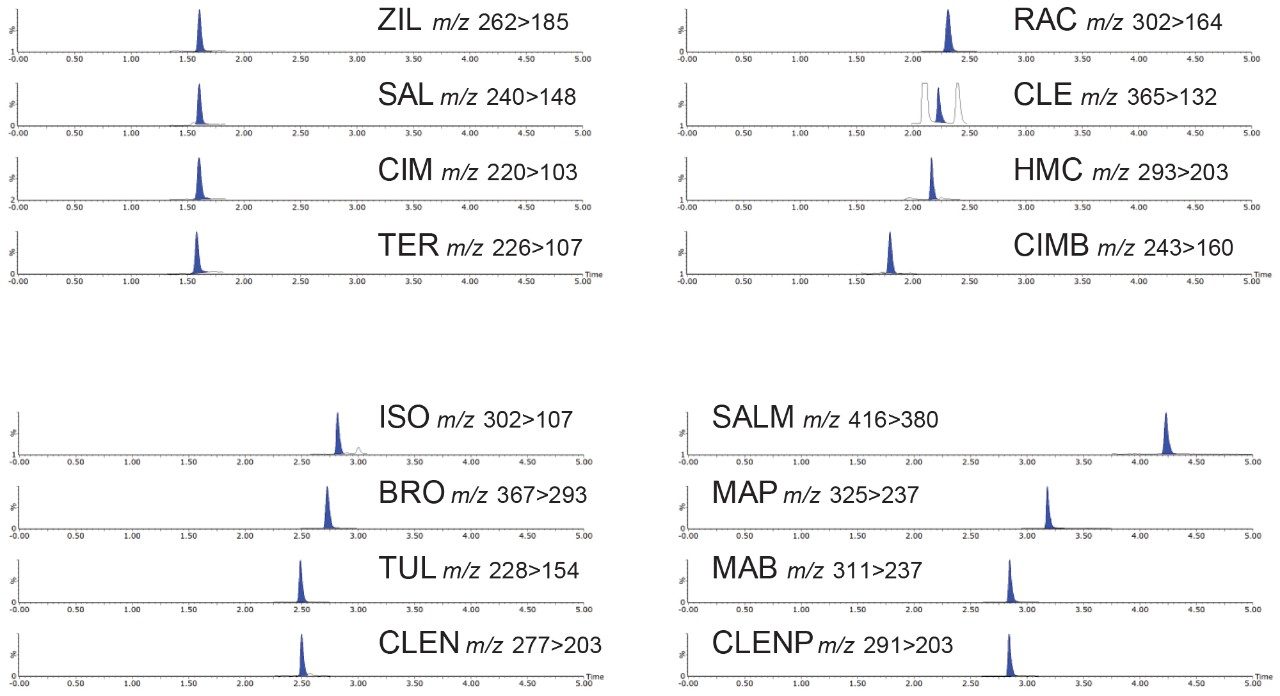
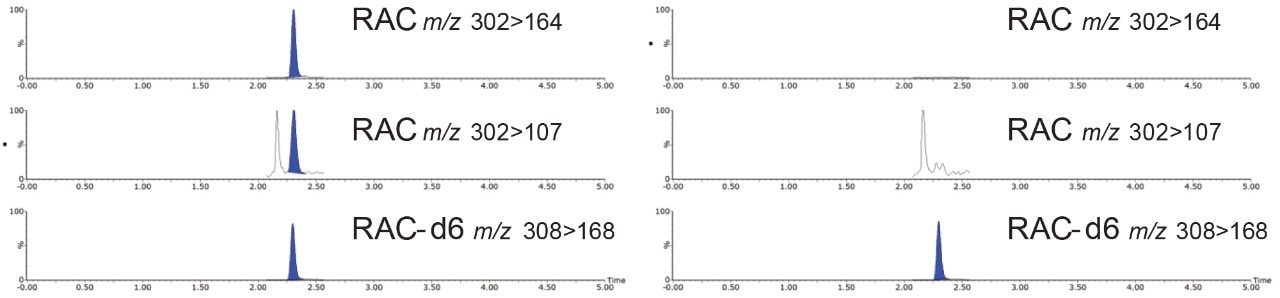
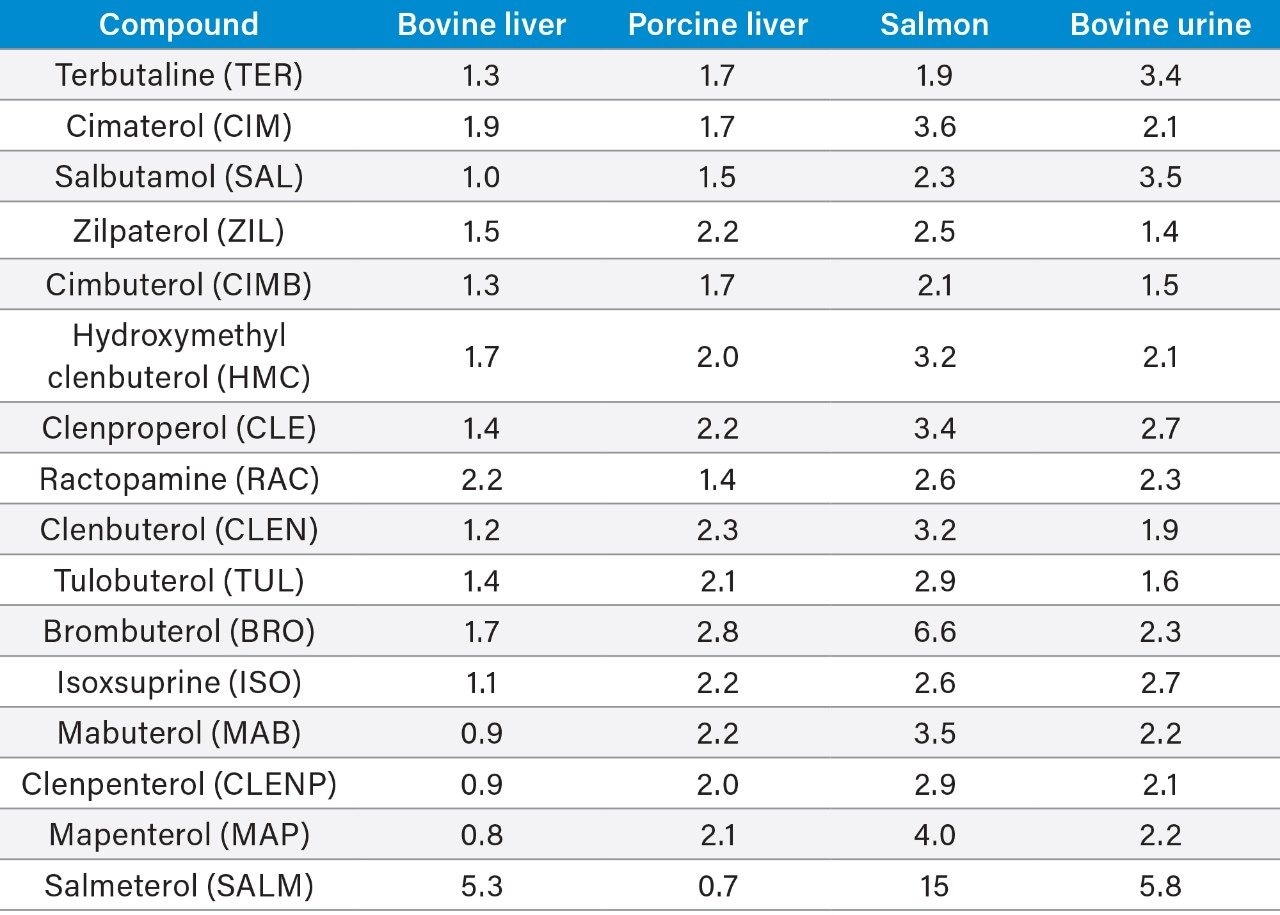
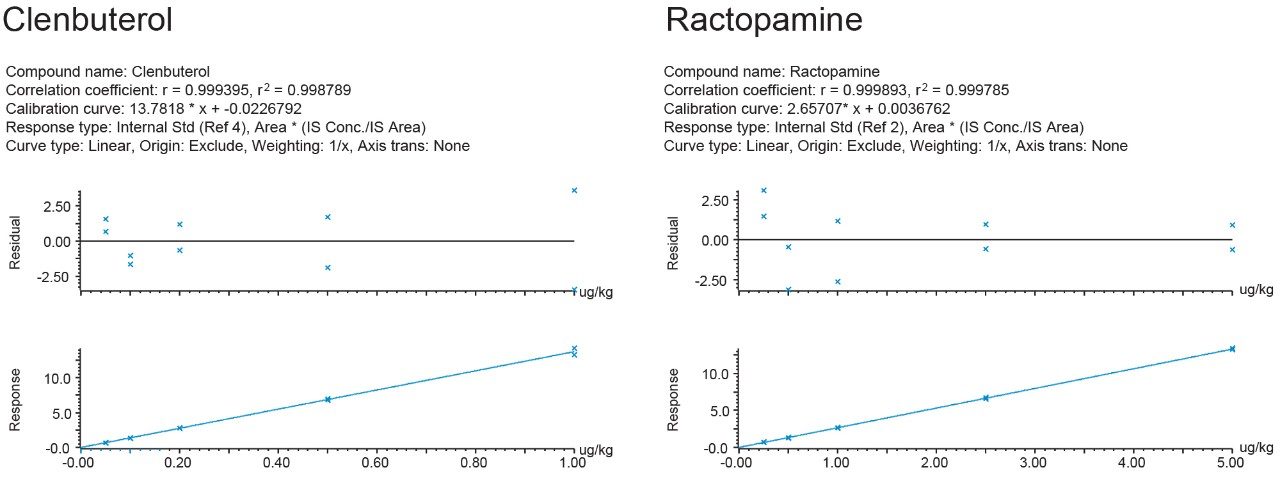
Most countries have established strict surveillance programs for official control purposes to check compliance with regulatory limits, for both domestic and imported produce. Monitoring compliance within these limits requires the use of highly sensitive and selective analytical methodology based on liquid chromatography-tandem mass spectrometry (LC-MS/MS). As the frequency of detection of residues is typically low, samples tend to be screened using a reduced but adequate level of quality control. This includes the use of stable isotope analogs as surrogates to monitor and mitigate the impact of any sample-to-sample variability in matrix effects. Any suspect positives are re-analyzed with the same method but with additional analytical quality control suitable for quantification (e.g. multi-level matrix-extracted calibration). This method had also been validated as suitable for confirmation4 with values for Decision Limit (CCα) as low as reasonably achievable.