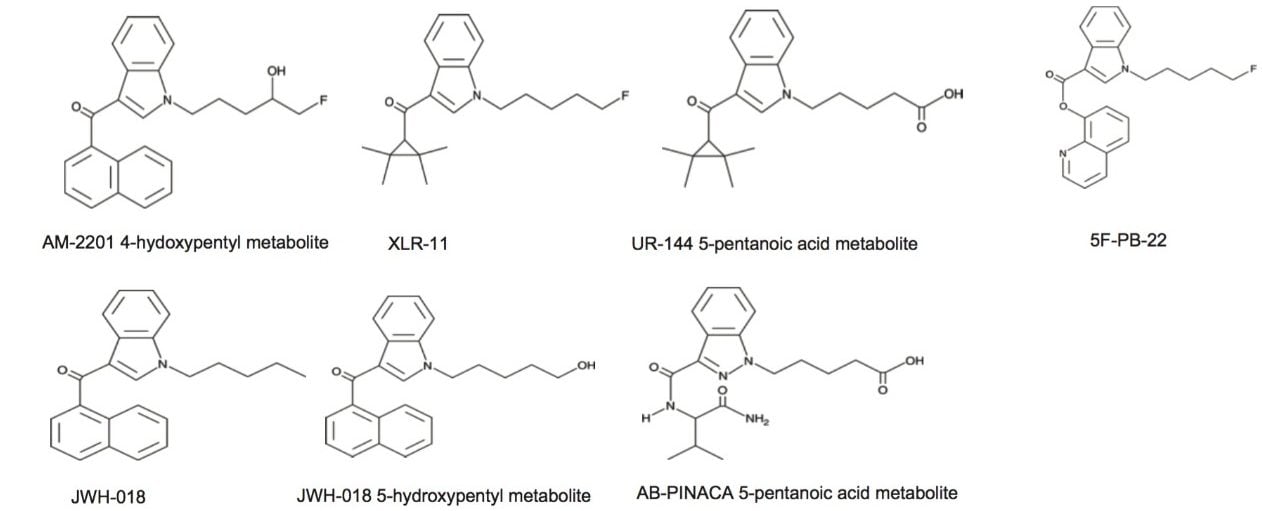
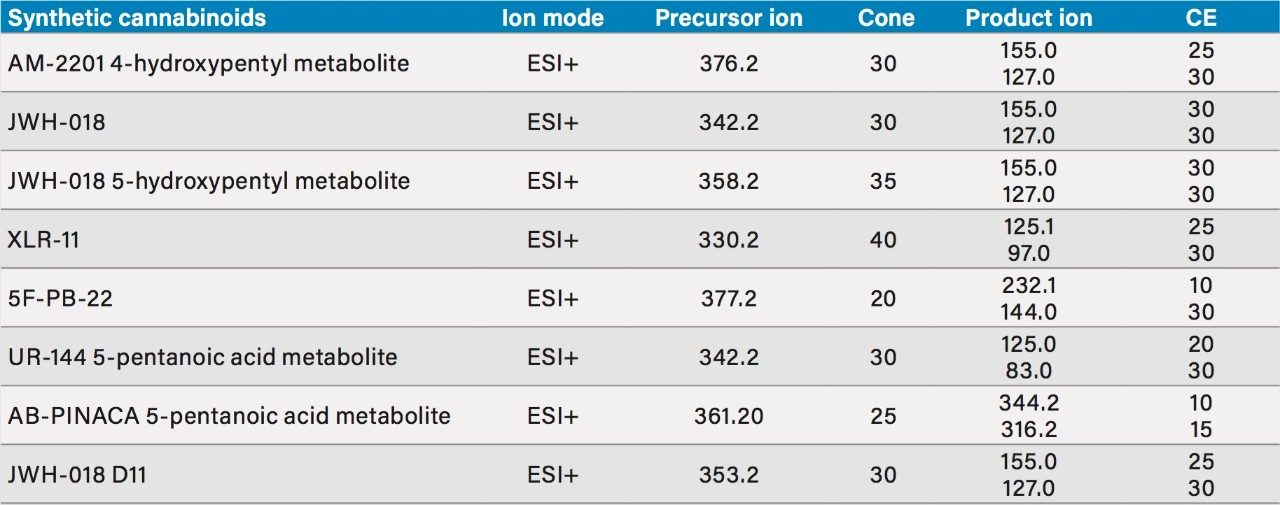
As stated by the Forensic Toxicology Council,1 the field of forensic toxicology deals with cases where drugs and/or chemicals have led to a death situation. An important function of the field relates to death investigation toxicology, also known as postmortem toxicology analysis. Since results are likely to be used in a court of law, therefore the accuracy and precision of the analytical technique is an essential prerequisite.
The core focus of a forensic toxicology laboratory is to determine the presence or absence of drugs in seized evidence. The target matrix can be in solid format, such as pills and powders, or biological materials (blood, plasma, urine, saliva, hair, and tissues). The analytical techniques currently available are divided into two categories, some platforms are used for screening methods (qualitative) and other solutions are used for confirmation methods (quantitative). Most laboratories are usually equipped with gas chromatography (GC) or liquid chromatography (LC) hyphenated to a mass spectrometer (MS). For several decades, GC/MS was the tool of choice for bio-analysis. However, with the introduction of atmospheric pressure ionization technique, LC/MS is now the most popular technique in the field of forensic toxicology.
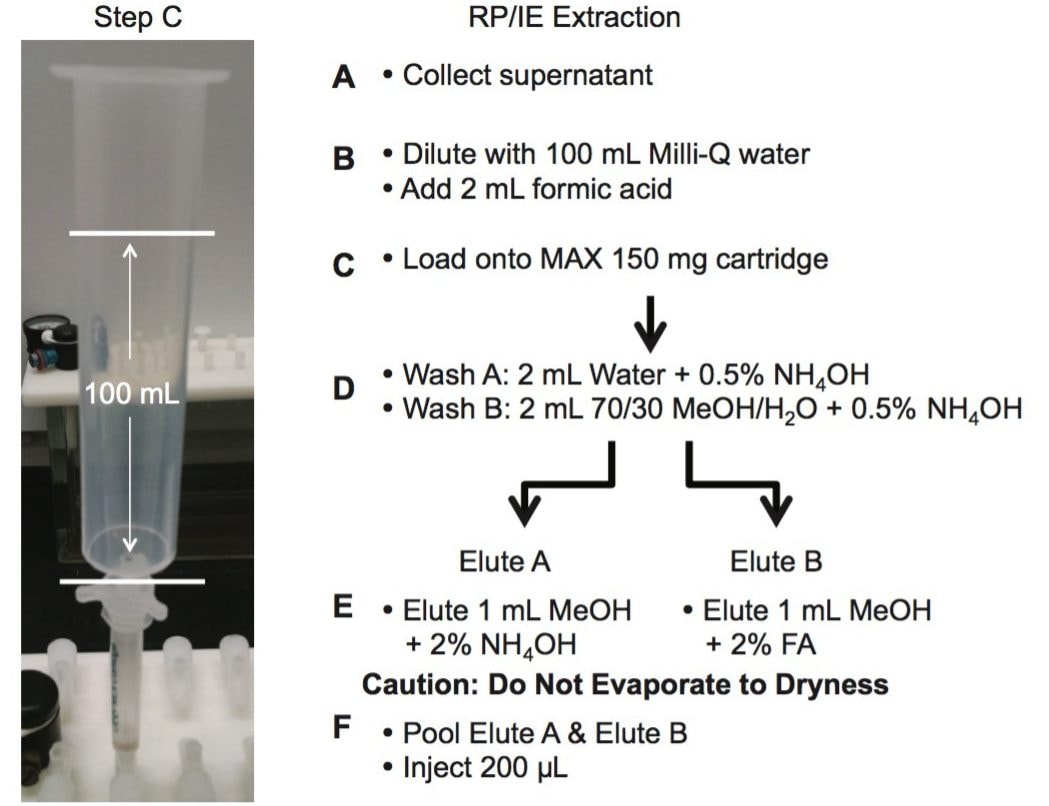
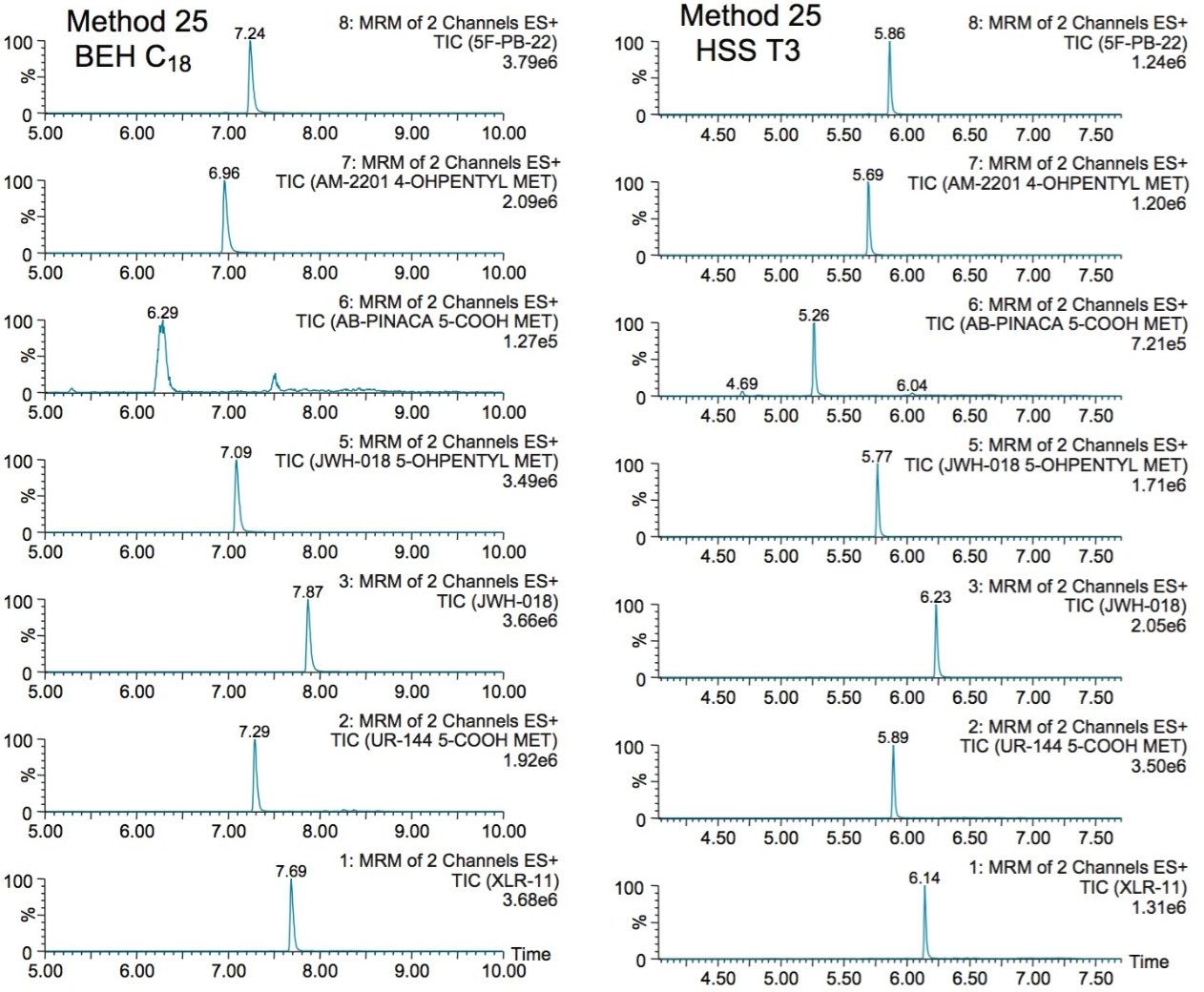
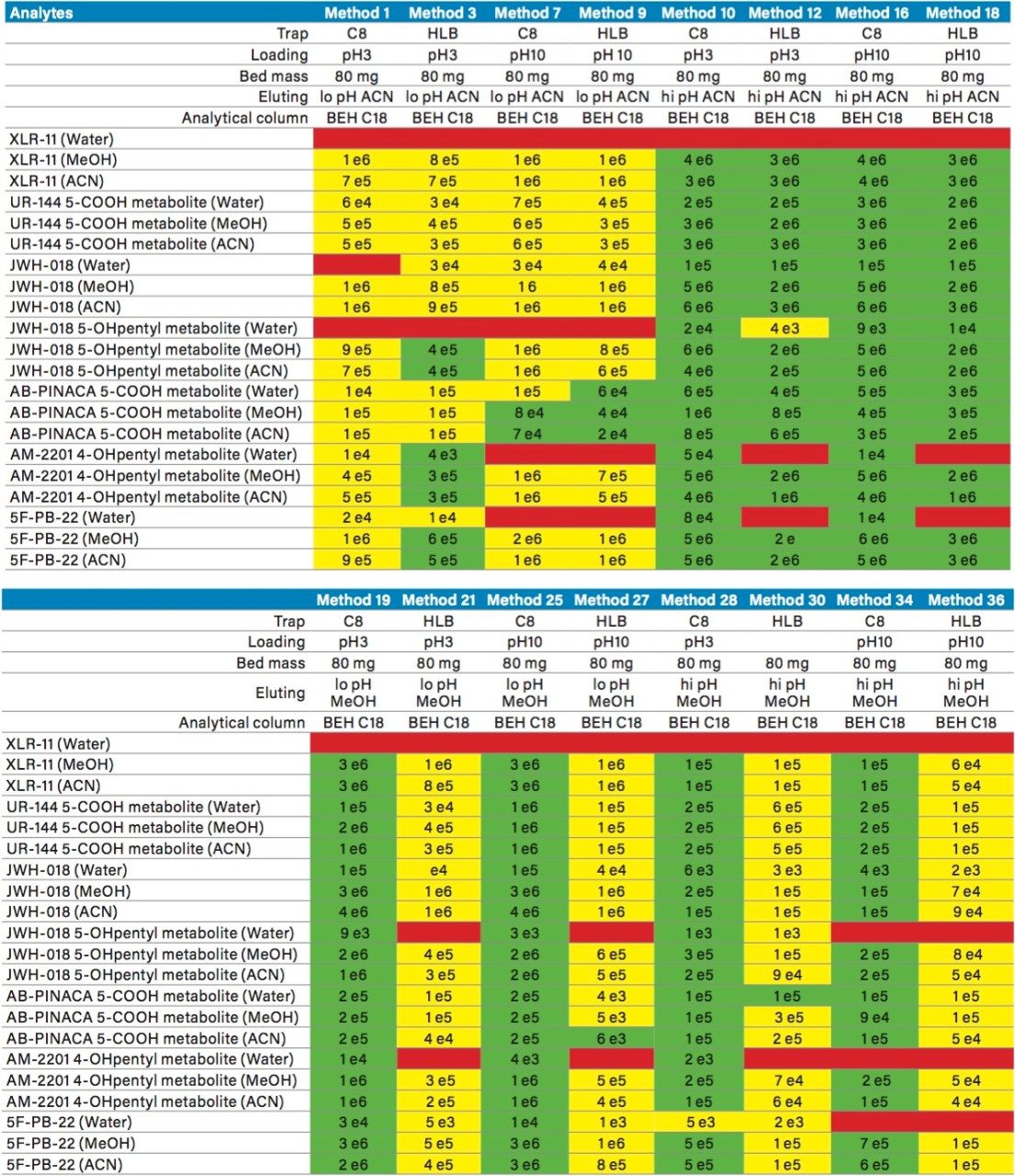
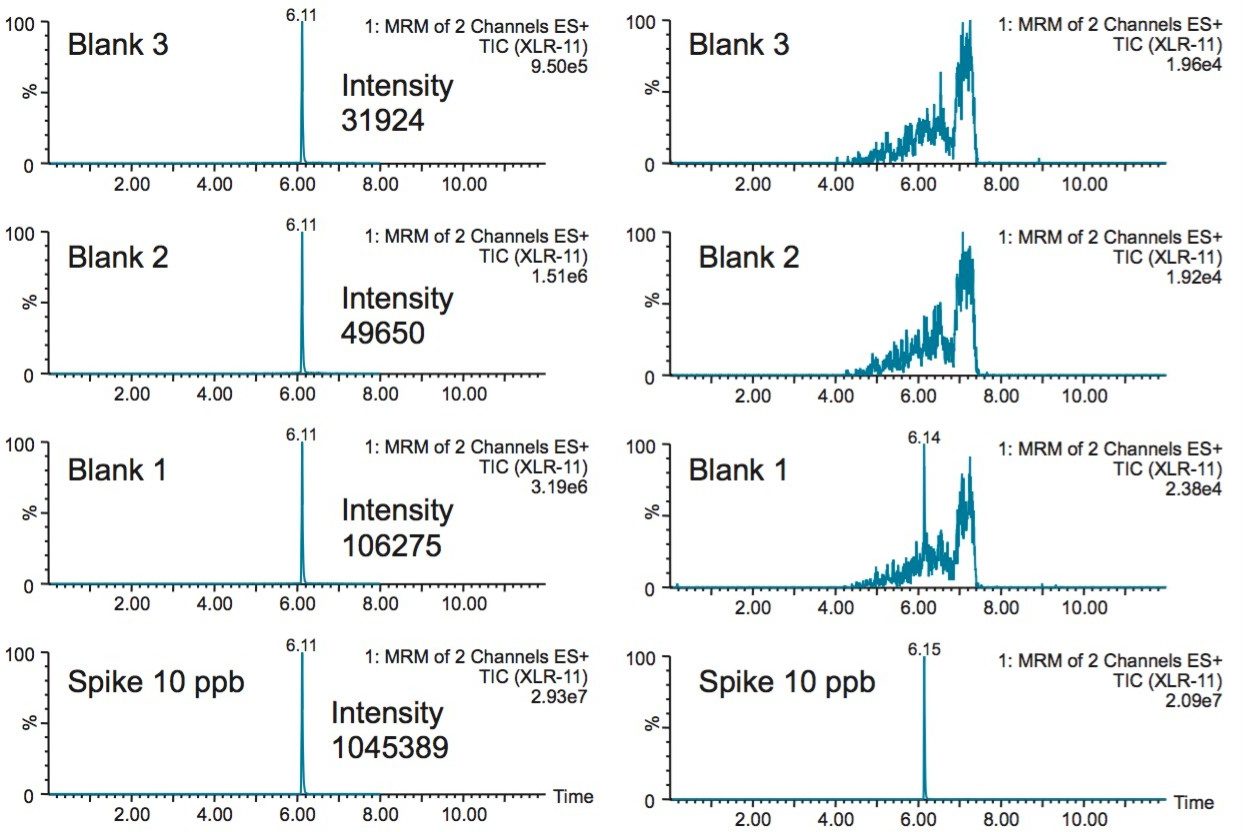
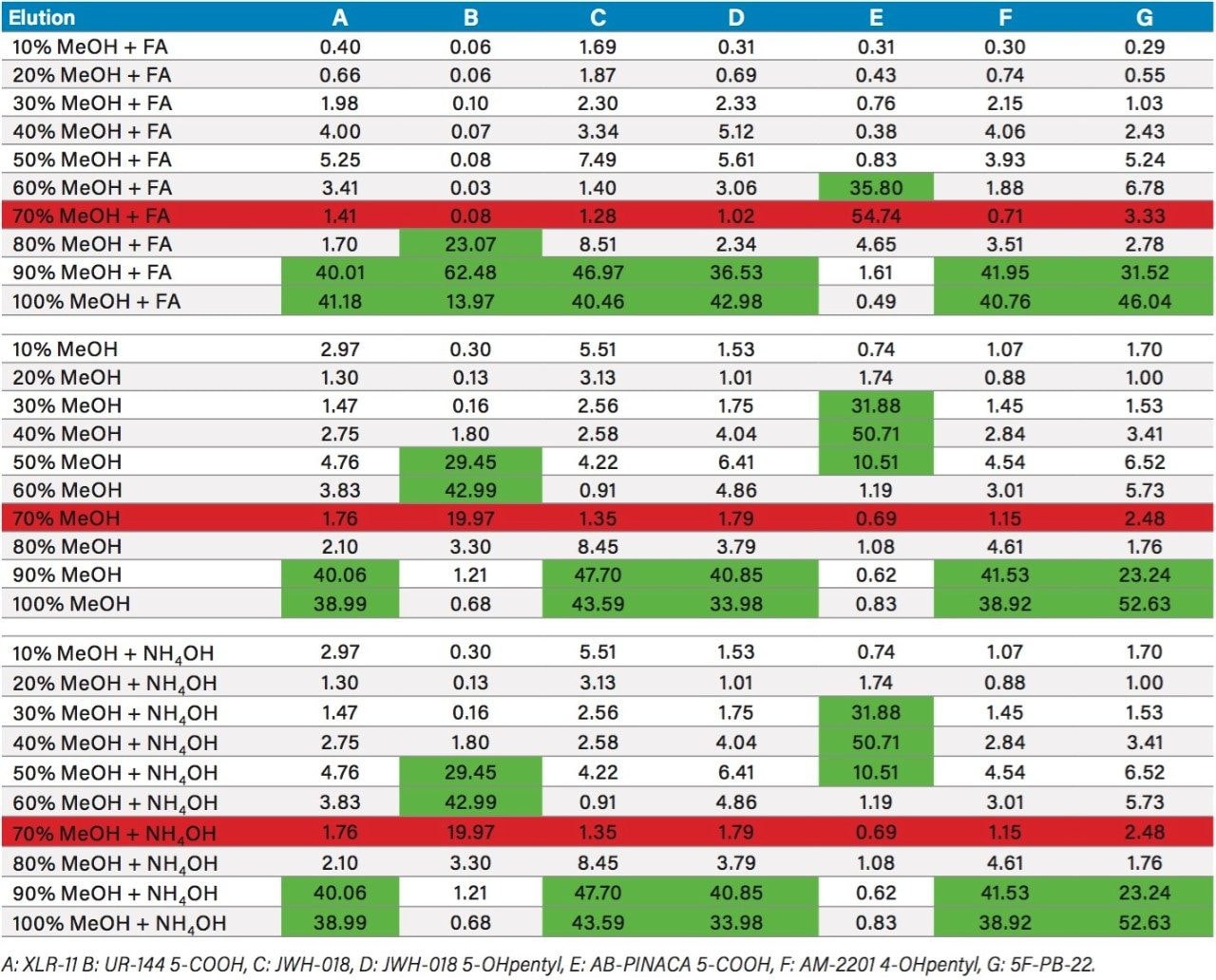
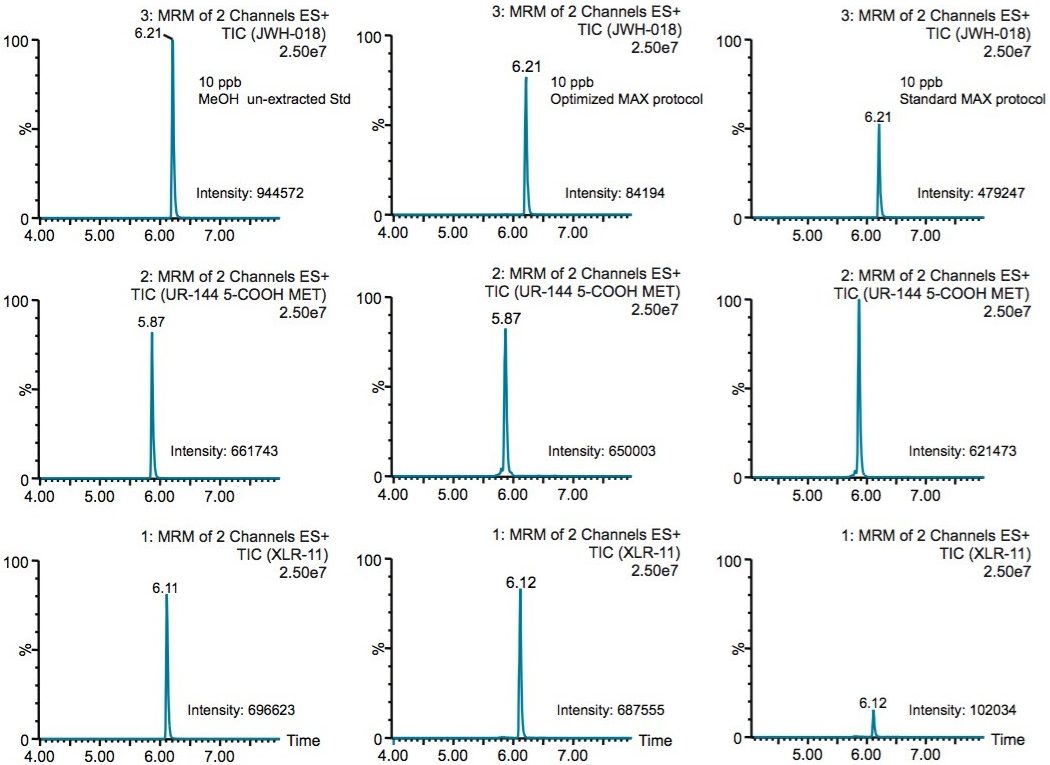
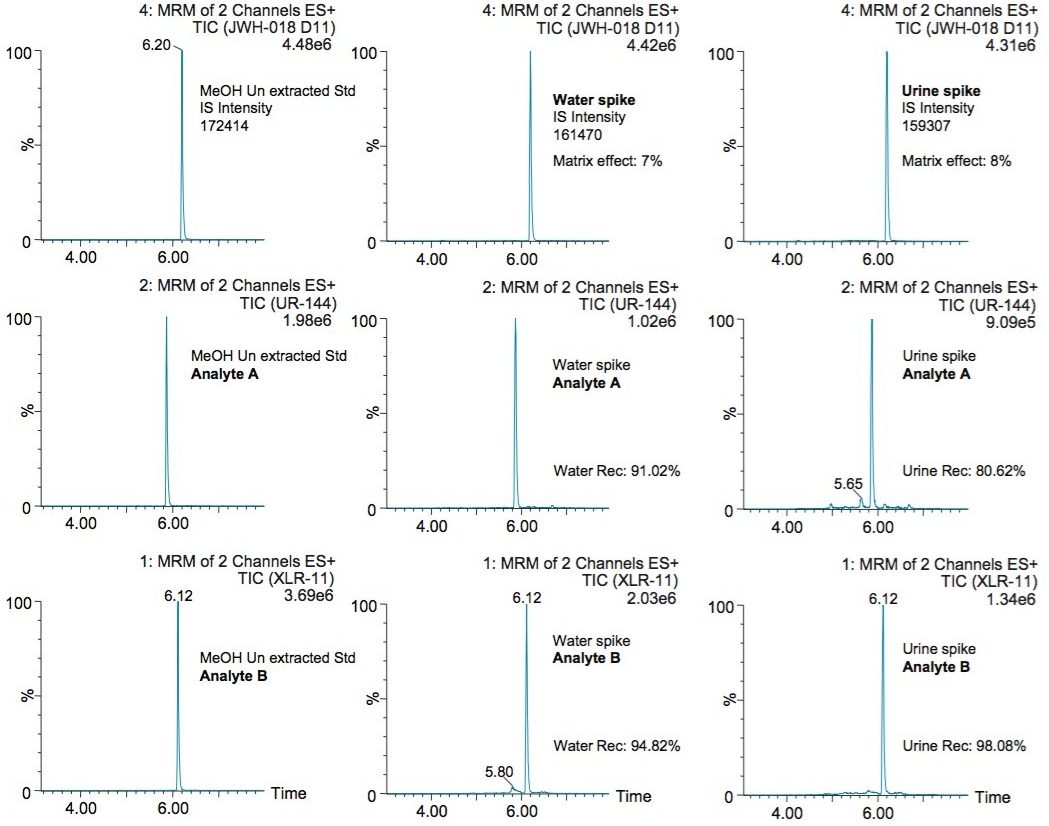
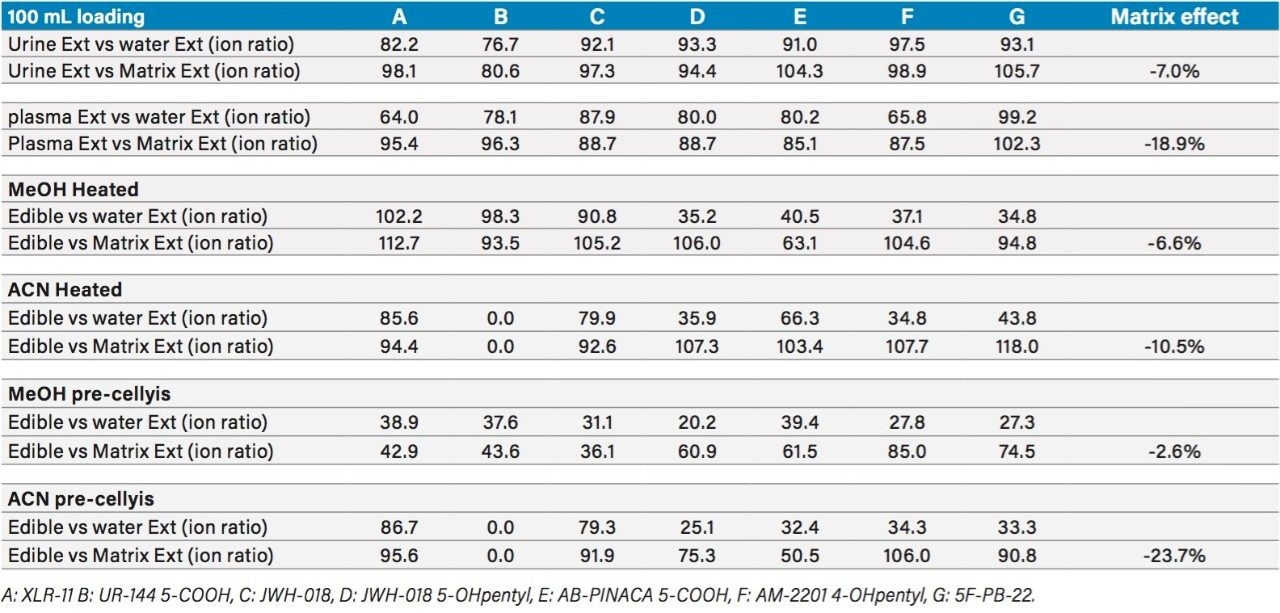
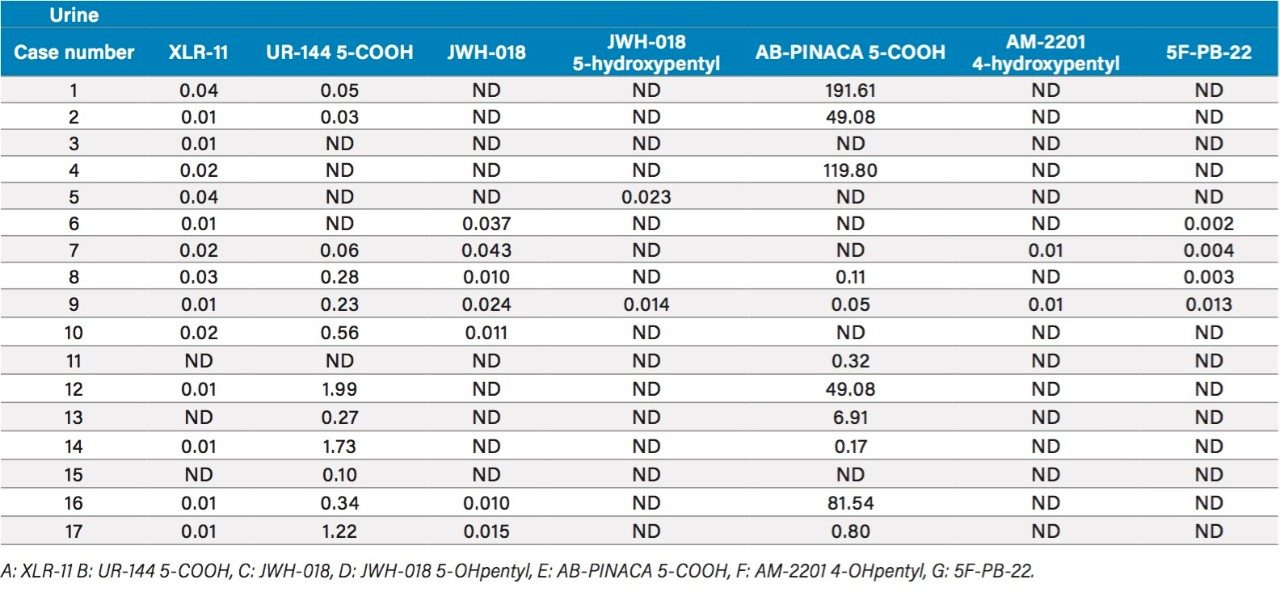
Detection and quantification of drugs in complex matrices are difficult to accomplish due to time-consuming extraction processes as well as the difficulty to detect analytes at trace levels. A robust extraction and clean up methodology, in which a homogenization step precedes, is a must in order to reach a targeted limit of detection (LOD) as well as maintain robust and reliable instrument performance. The use of advanced hyphenated instrumentation platforms, such as UPLC-MS/MS, has allowed analysts to detect trace levels of analytes.
Traditional extraction techniques used in most laboratories are decades old and do not have the robustness to produce quality results. A novel micro extraction protocol, combined with a multi-dimension chromatography (2D LC-MS/MS), is described within, resulting in a decreased sample preparation time without sacrificing the quality of results often observed when utilizing current single dimension chromatography techniques.2,3,4