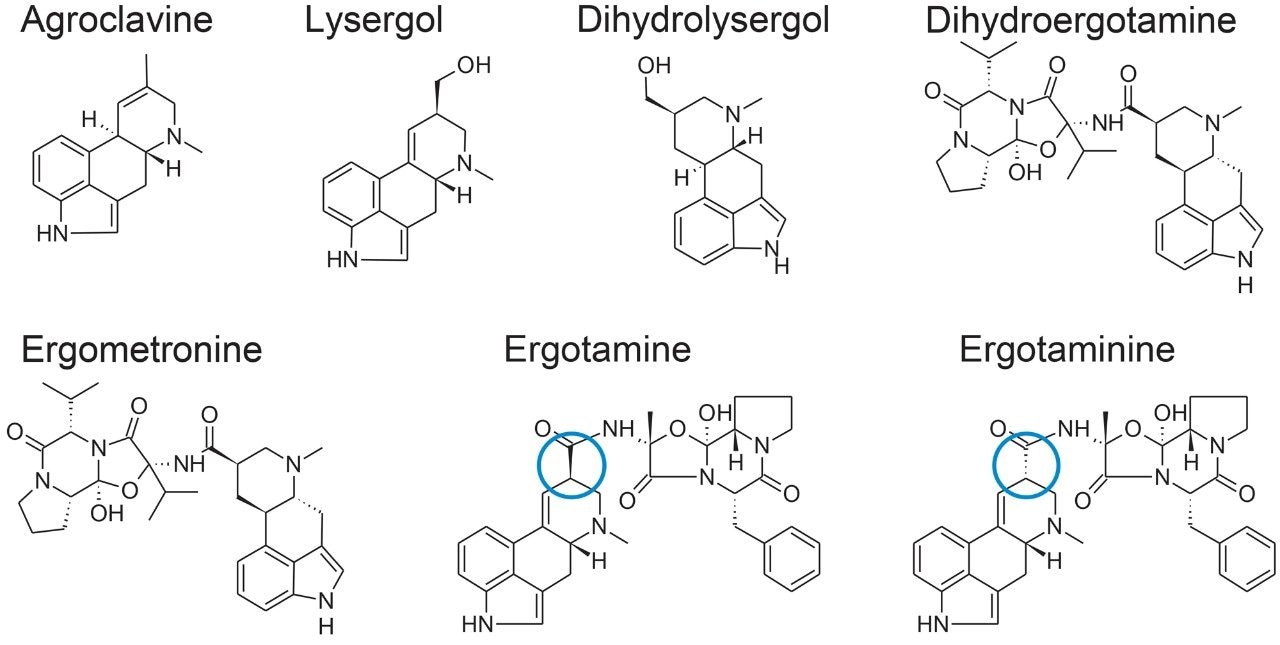
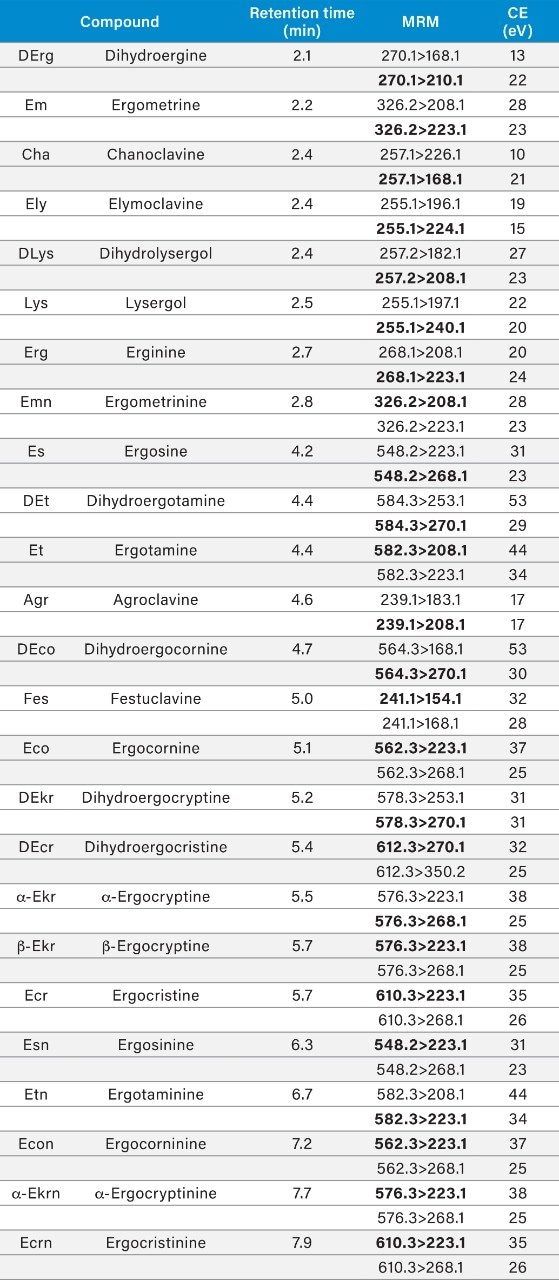
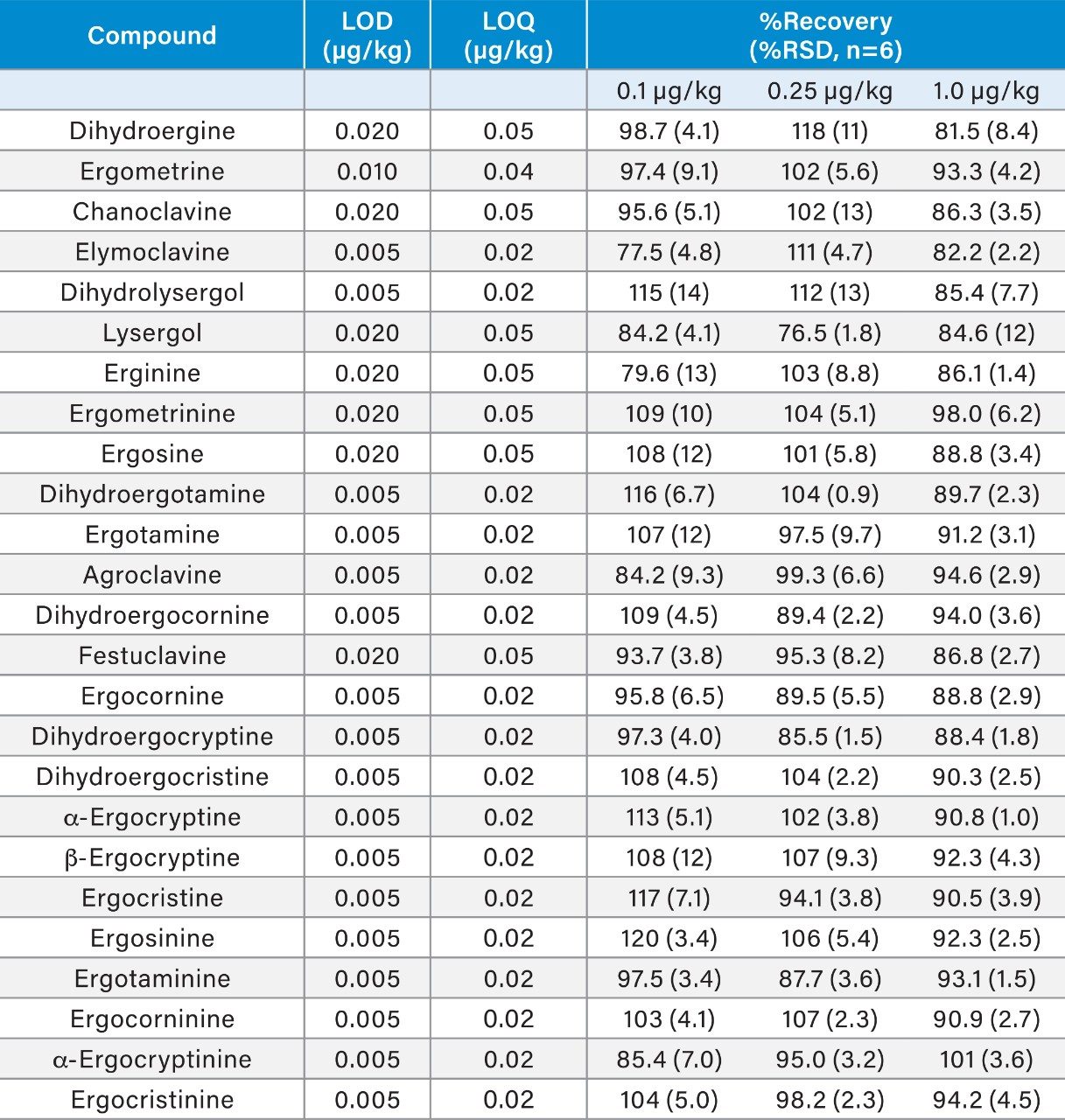
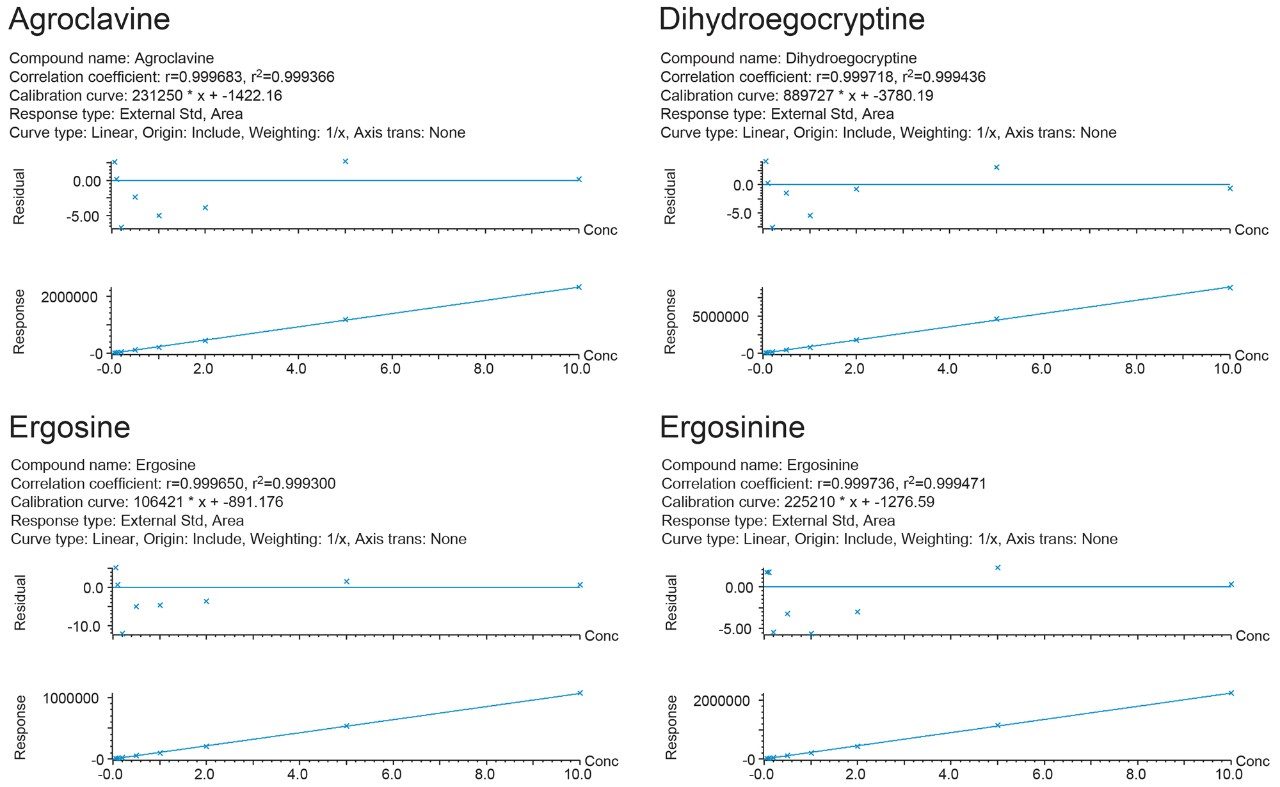
Ergot alkaloids (EAs) are mycotoxins produced by fungi including Claviceps spp. Among the cereal species, rye and triticale that have open florets are known to be especially susceptible, but wheat, barley, oats, and other cereal grains are also potential fungal hosts.1 The fungus replaces the developing grain or seed with the alkaloid-containing wintering body, known as the ergot body or sclerotium. The sclerotia are harvested together with the cereals or grass and can thus lead to contamination of cereal-based food and feed products with EAs. Ingestion of contaminated products can cause a number of harmful effects in humans and livestock. Although ergotism has practically been eliminated as a human disease, it remains an important veterinary problem, particularly in cattle, horses, sheep, pigs, and chicken.2 There are three main classes of EAs: short chain substituted amides of lysergic acid, clavine alkaloids, and ergopeptines, which are peptide EAs comprising (+)-lysergic acid and a tripeptide system containing L-proline. Structures of some of the major EAs are shown in Figure 1, including an example of epimerisation of an ergopeptine at the C8 position to form the corresponding ergopeptinine. Although the ergopeptinines are described as biologically inactive, interconversion can occur under various conditions so analytical methods should include the determination of both epimeric forms.