Mutagenic impurities, formerly known as genotoxic (GI) or potential genotoxic impurities (PGI), are compounds that have the potential to modify DNA, and as a consequence can cause cancer. It is important that impurities potentially present in the marketed drug are evaluated early in the drug development process. To that end, analytical methods must be developed that are sensitive and specific enough to determine the levels in both drug substance and product.
The International Conference on Harmonization published ICH M7 guidelines, which highlight the requirements for assessment and control of DNA-reactive impurities to ensure the safety of pharmaceutical products.1 The European Medicine Evaluation Agency (EMEA), U.S. FDA, and the Asia regulatory agencies all follow these guidelines. They require that any mutagenic impurities in a drug substance or drug product must be below the Threshold of Toxicological Concern (TTC) of 1.5 µg per day based upon the maximum daily dosage of the pharmaceutical compound over a lifetime. For example, for a dosage of 1 g of Active Pharmaceutical Ingredient (API) per day, any impurity must be less than 1.5 ppm (1.5 µg). This is orders of magnitude lower than for general pharmaceutical impurities analysis, which is at the 500 ppm level and governed by Q3B(R).2
Pharmaceutical analysis is typically performed using LC with UV detection for non-volatile compounds, or GC with FID detection for volatile compounds. However, the low levels of detection required for mutagenic impurities present a significant challenge. In these situations, MS detection is required in order to achieve the desired sensitivity. Some of these methods are required to provide support during the whole life cycle of a drug from early development through to manufacturing quality control. Typical reverse-phase (RP) (where the majority of separations are done on C18 stationary phases) and normal-phase (NP) chemistries can be used; this opens up a wide range of selectivity choices to help develop successful separations.
Convergence chromatography (CC) is a chromatographic technique similar to HPLC, but instead of the weak mobile phase being aqueous it is replaced with supercritical carbon dioxide (CO2). Supercritical CO2 can be paired with a large number of different co-solvents to increase the solvating power. CO2 is miscible with the whole range of the eluotropic series opening up a large choice of solvent selectivity – with methanol, IPA, ethanol, and acetonitrile being the most commonly used co-solvents.
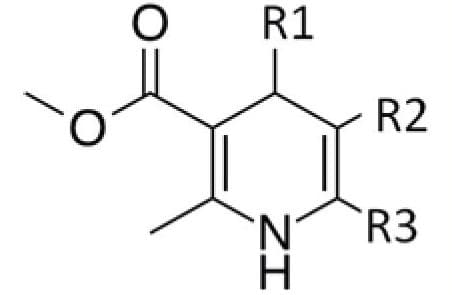
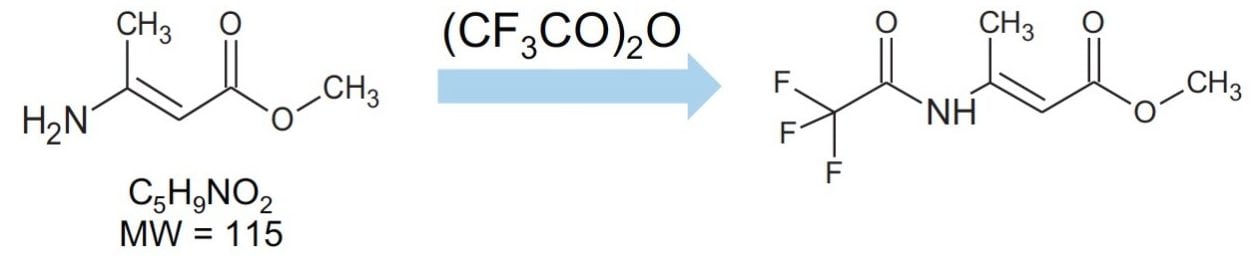
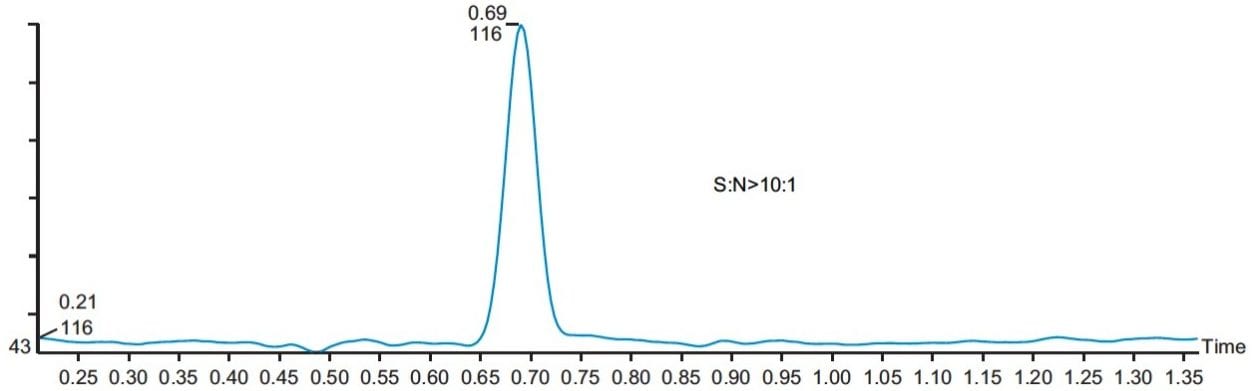
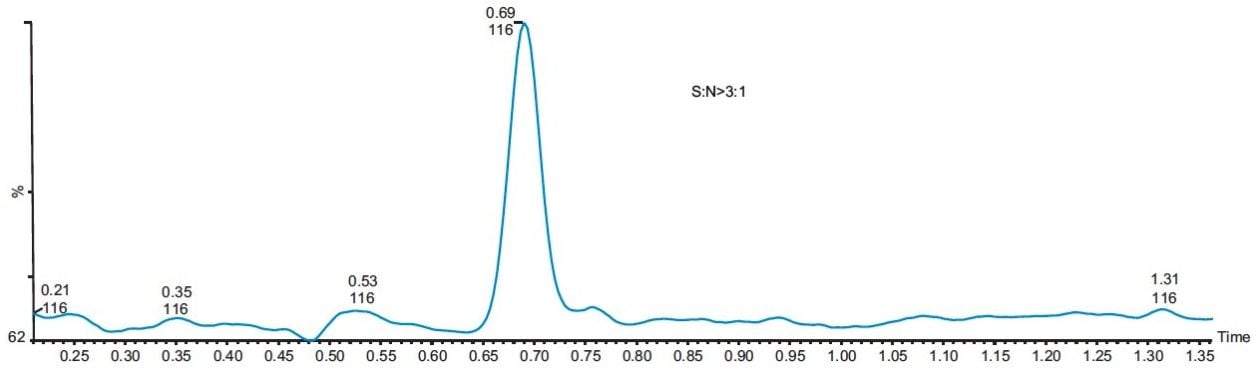
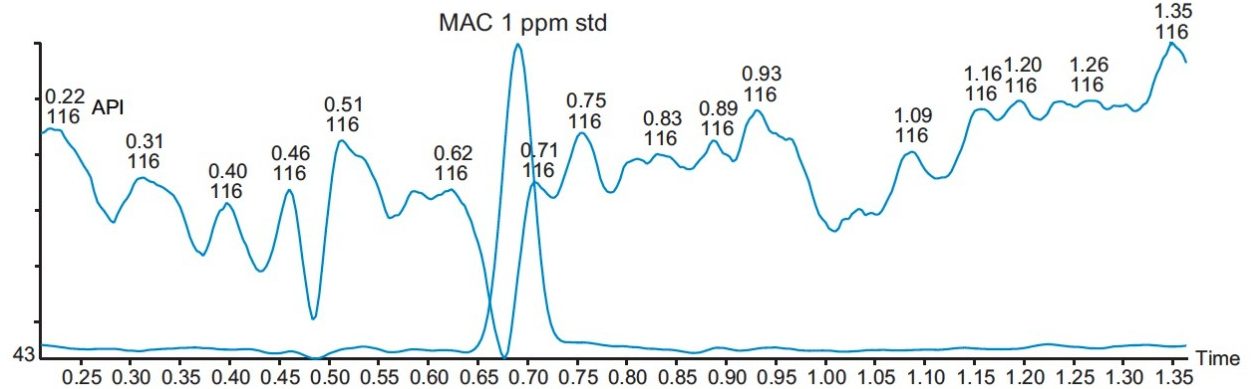
Methyl-3-aminocrotonate (MAC) is a Michael-reactive receptor and a starting material in a number of different cardiovascular drug products. The API used is an active substance from a proprietary drug product; therefore, only the partial structure is shown in Figure 1. MAC flags up a positive from the mutagenic structural alerts. This compound would typically be analyzed by static head space (SHS) GC-MS after derivatization with trifluoroacetic anhydride to increase the volatility (Figure 2).3 When MAC (underivatized) was analyzed by UPLC-MS nothing was seen; this was thought to be due to its poor stability in aqueous solvents.
In this type of trace analysis where there is a large amount of matrix it would be advantageous if chemical derivatization of the mutagenic impurity can be avoided for the following reasons:
- The formation of acylation derivatives can be difficult to prepare
- Reaction bi-products can occur, which could add more complexity to the matrix
- The extra derivatization step would require extra validation to be completed
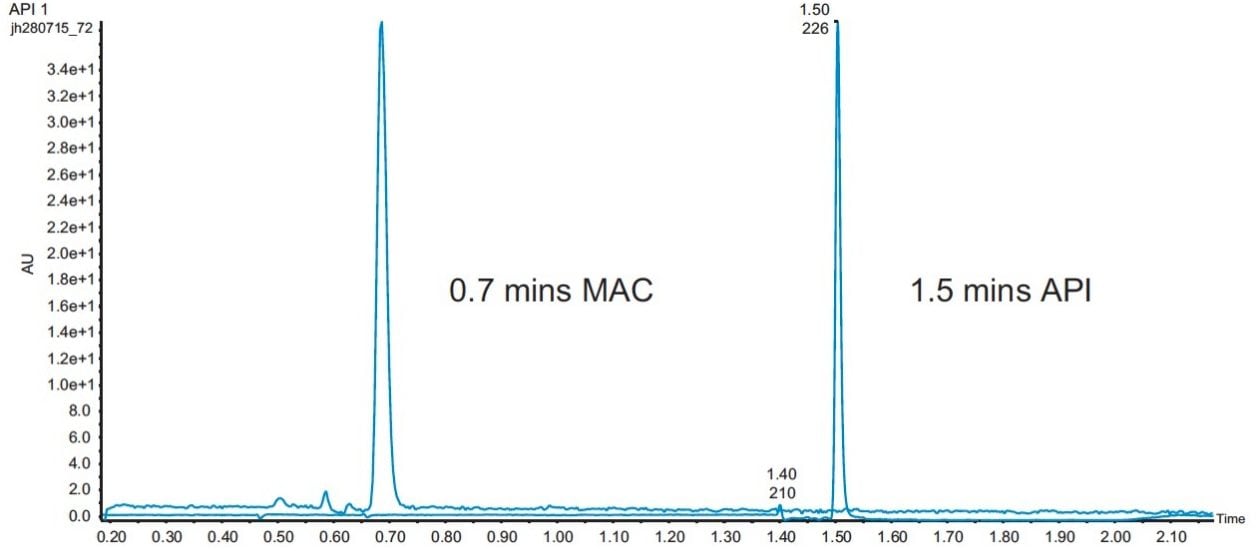
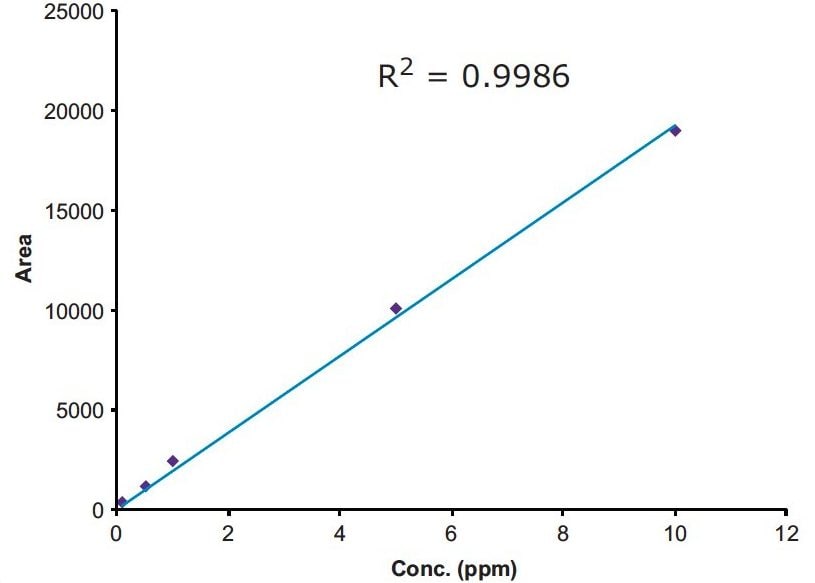
To avoid the above issues, this application note discusses the work carried out to investigate the use of Waters UltraPerformance Convergence Chromatography (UPC2) and MS detection using the ACQUITY QDa as an alternative technology for the analysis of MAC without the prior need of derivatization, for the detection and quantification in an active pharmaceutical ingredient (API).