The rise of biotherapeutics in the drug discovery and development pipeline has resulted in the increasing demand for protein/peptide bioanalysis by LC-MS techniques. This has led to a renewed interest in high resolution mass spectrometry (HRMS) platforms for use in quantitation because they enable both a larger mass range and additional options for selectivity. HRMS platforms retain the ability to quickly switch back to characterization mode to elucidate or resolve qualitative, underlying matrix issues with the assay. In this way, both peptide mapping and quantitation can be performed on the same platform.
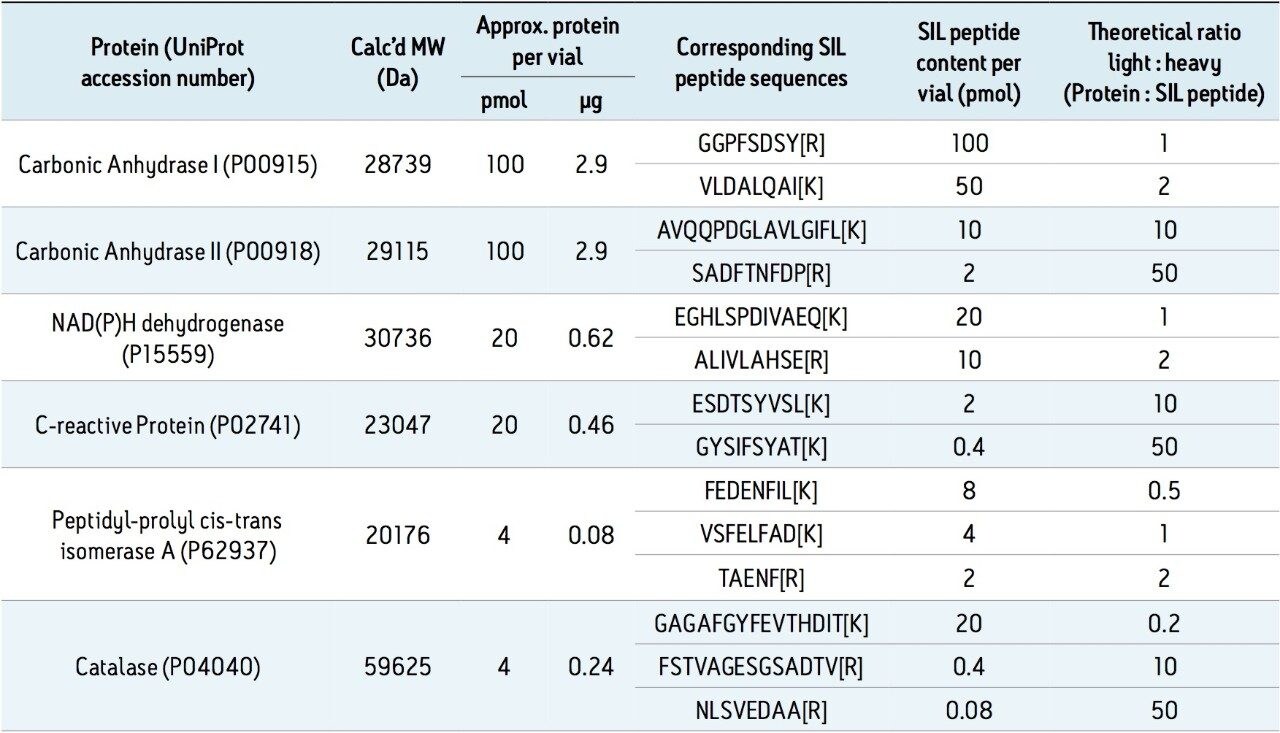
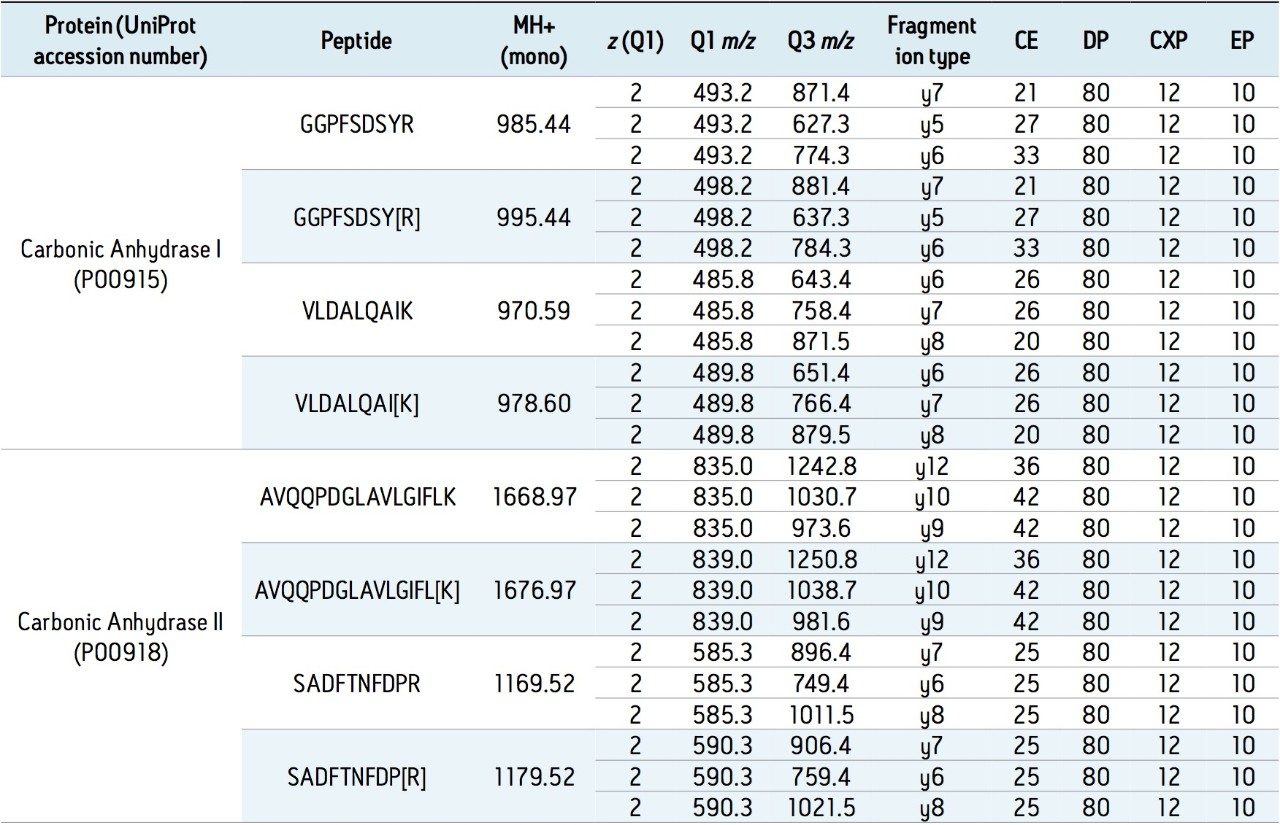
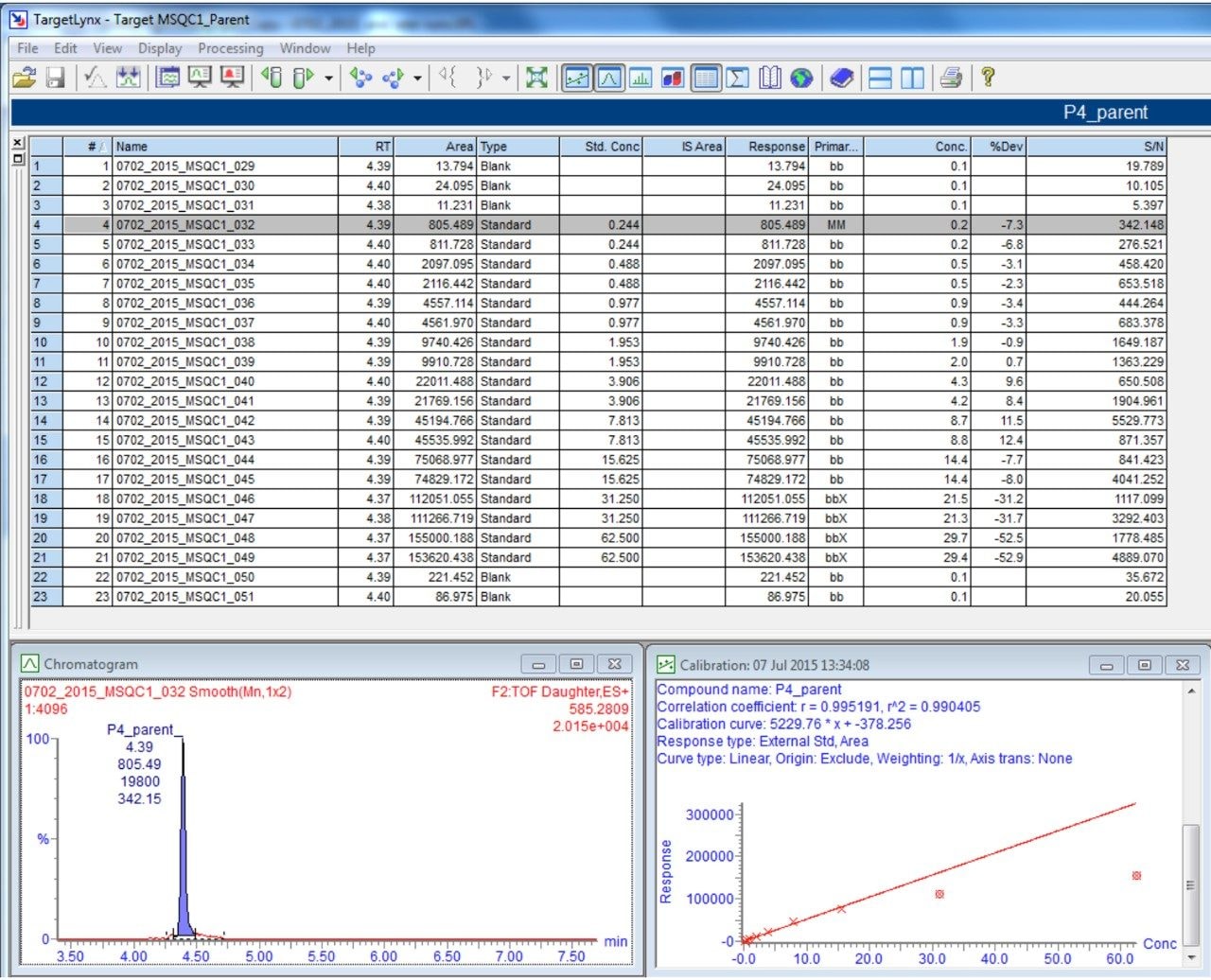
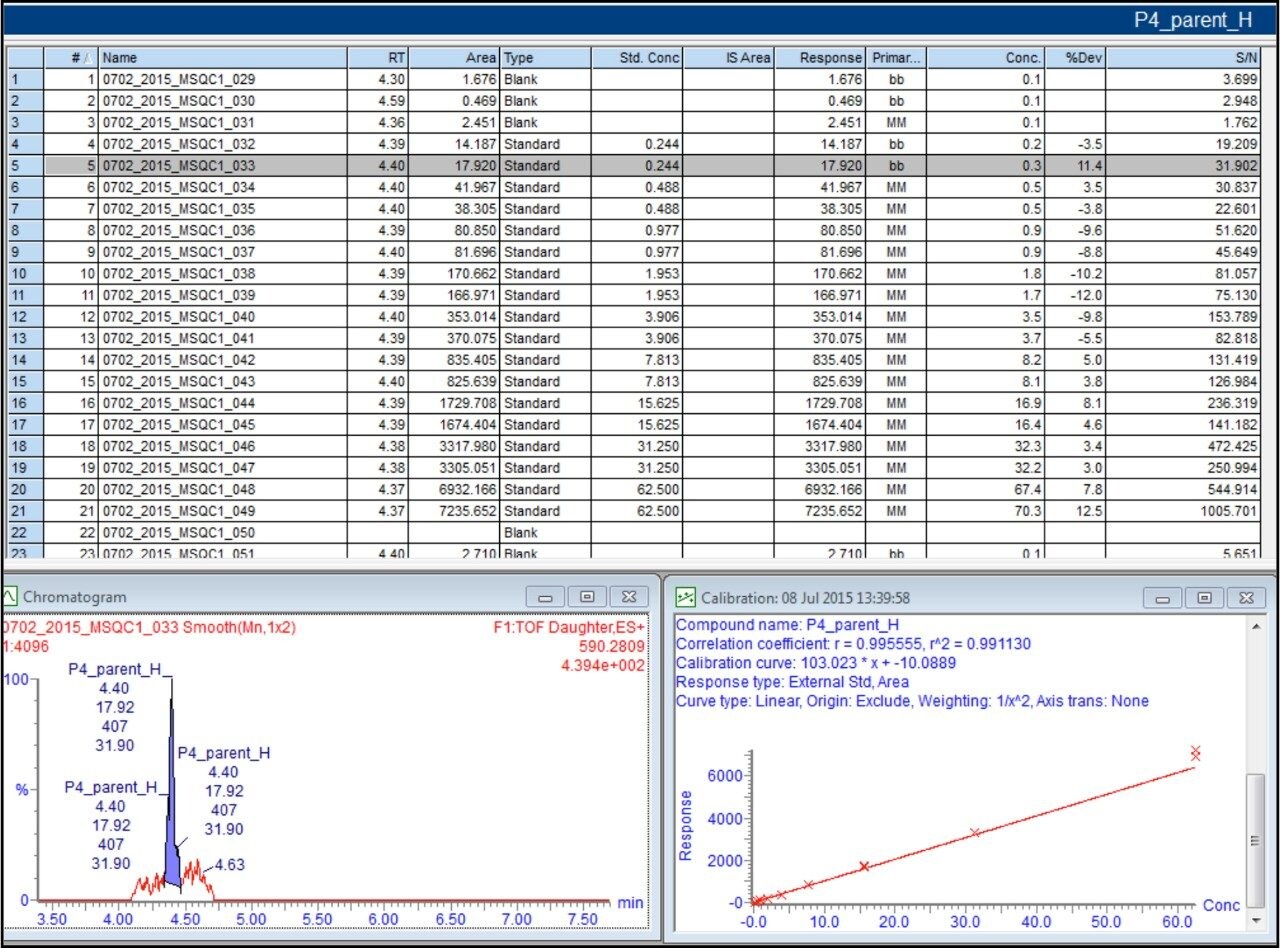
In addition to full scan data acquisition, newer targeted HRMS modes enable faster tandem-like throughput, while avoiding some of the complexity of full scan approaches. Currently, quantitation through a surrogate peptide approach remains the most popular practice for large molecule bioanalysis. In this approach, proteins and peptides are prepared through enzyme digestion. The resulting mixture is analyzed first to map out peptides thus formed and to search for signature peptides. Informatics tools such as Skyline are tailored for developing quantitative methods.
Once the signature peptides are identified, subsequent peptide quantitation is carried out using either tandem quadrupoles or high resolution mass spectrometers.
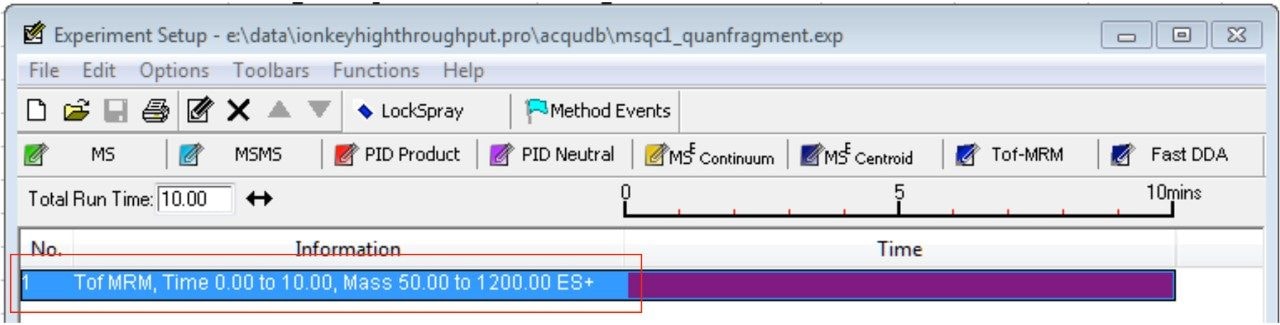
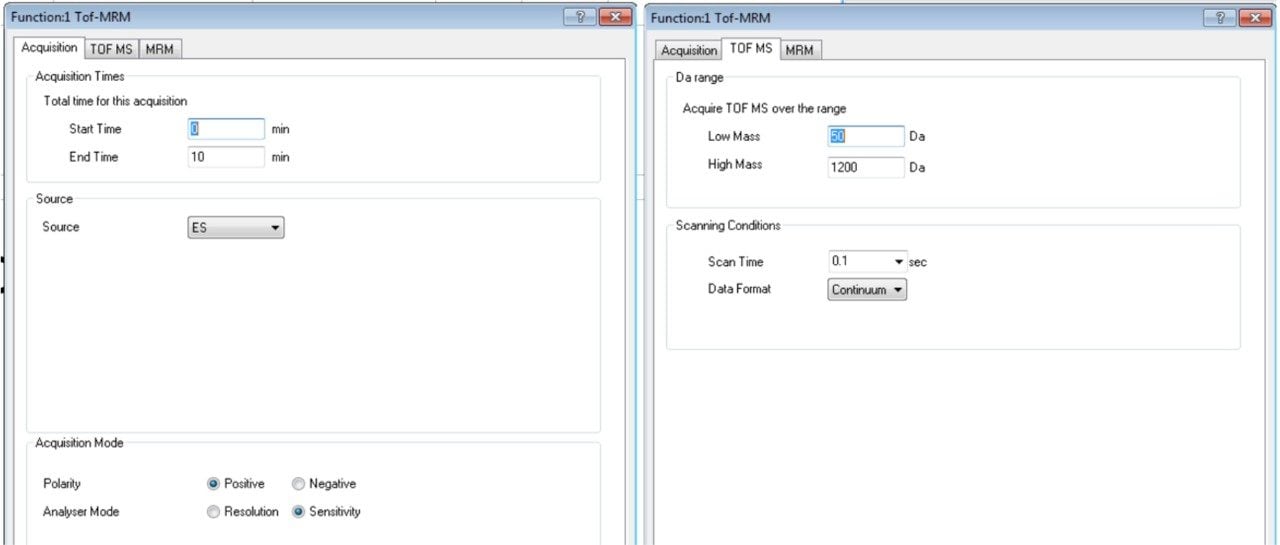
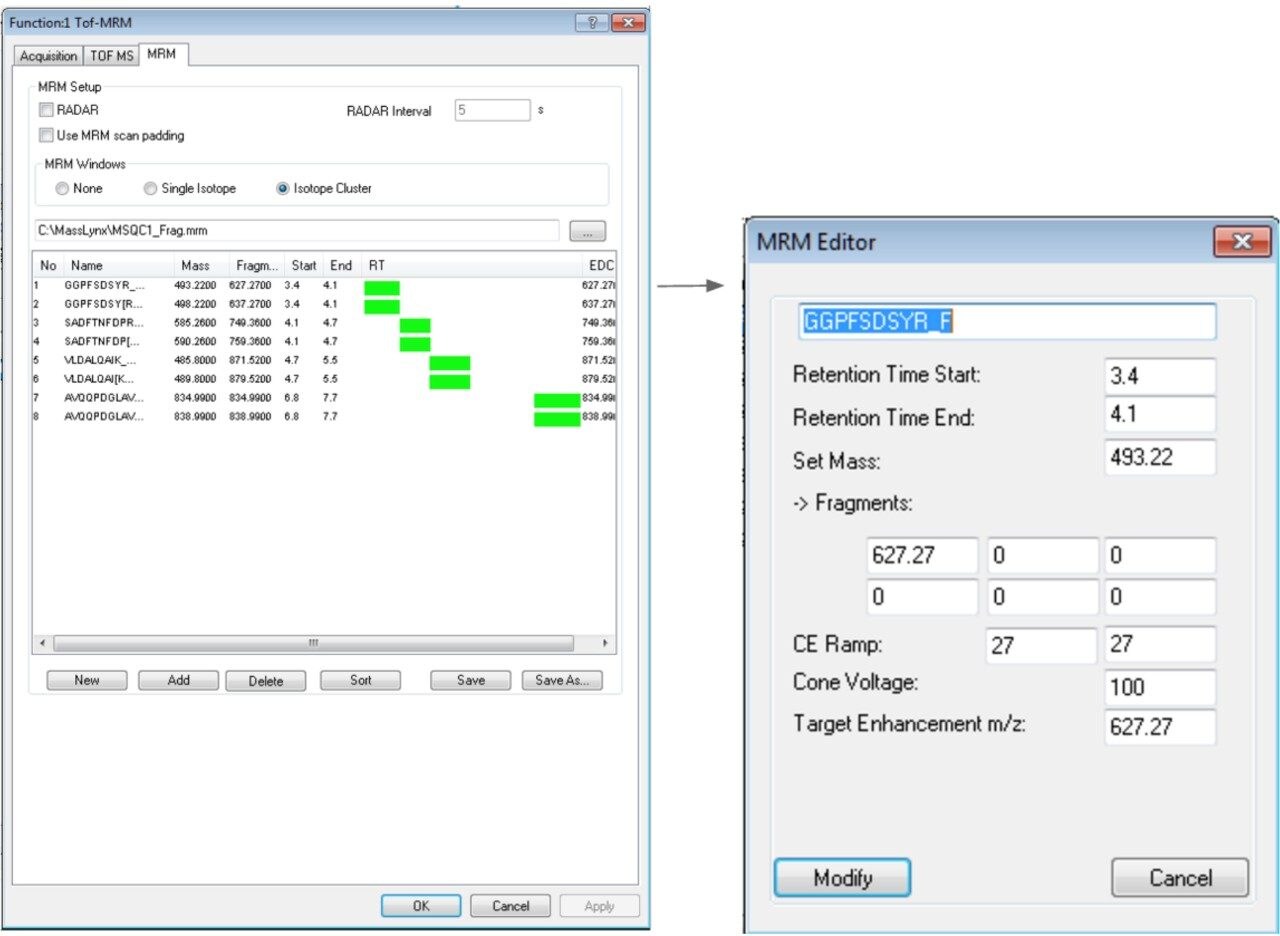
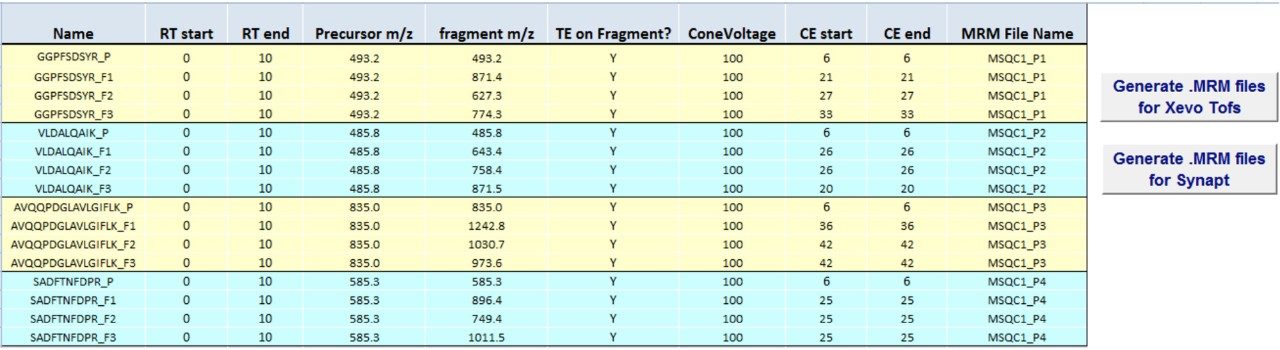
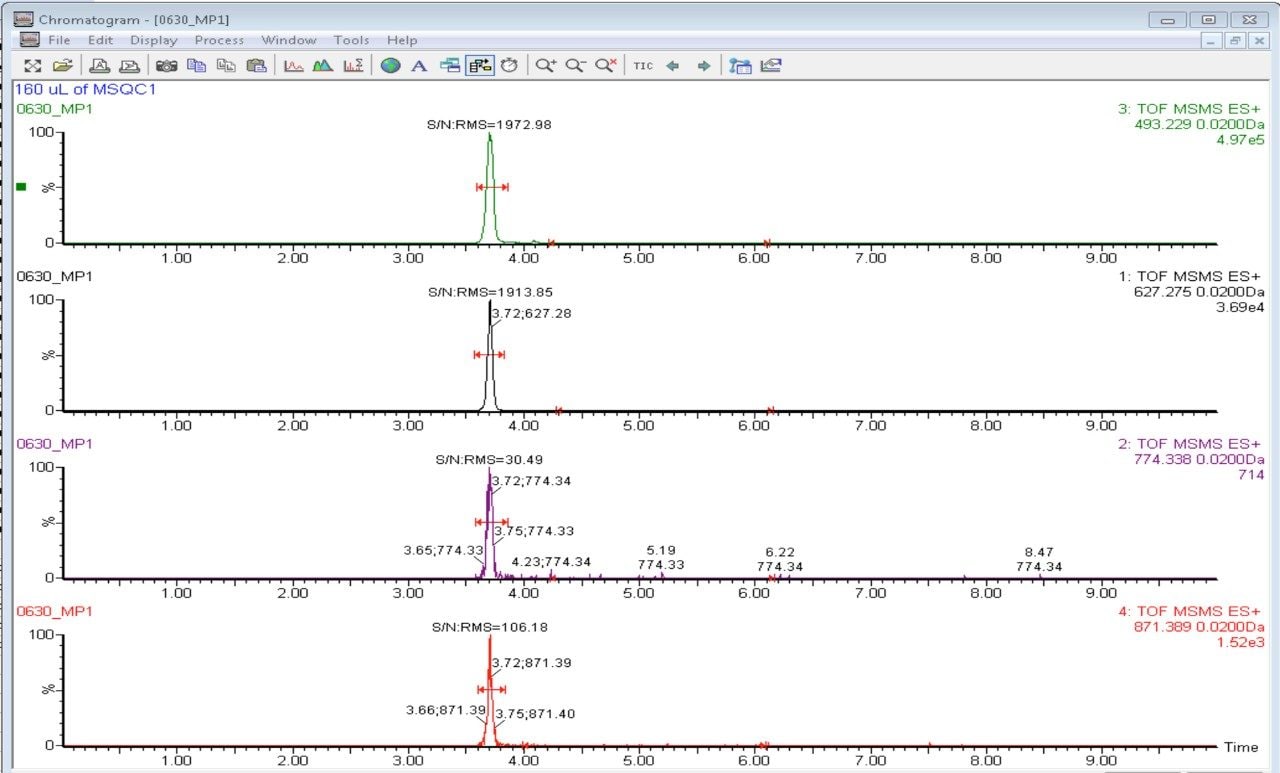
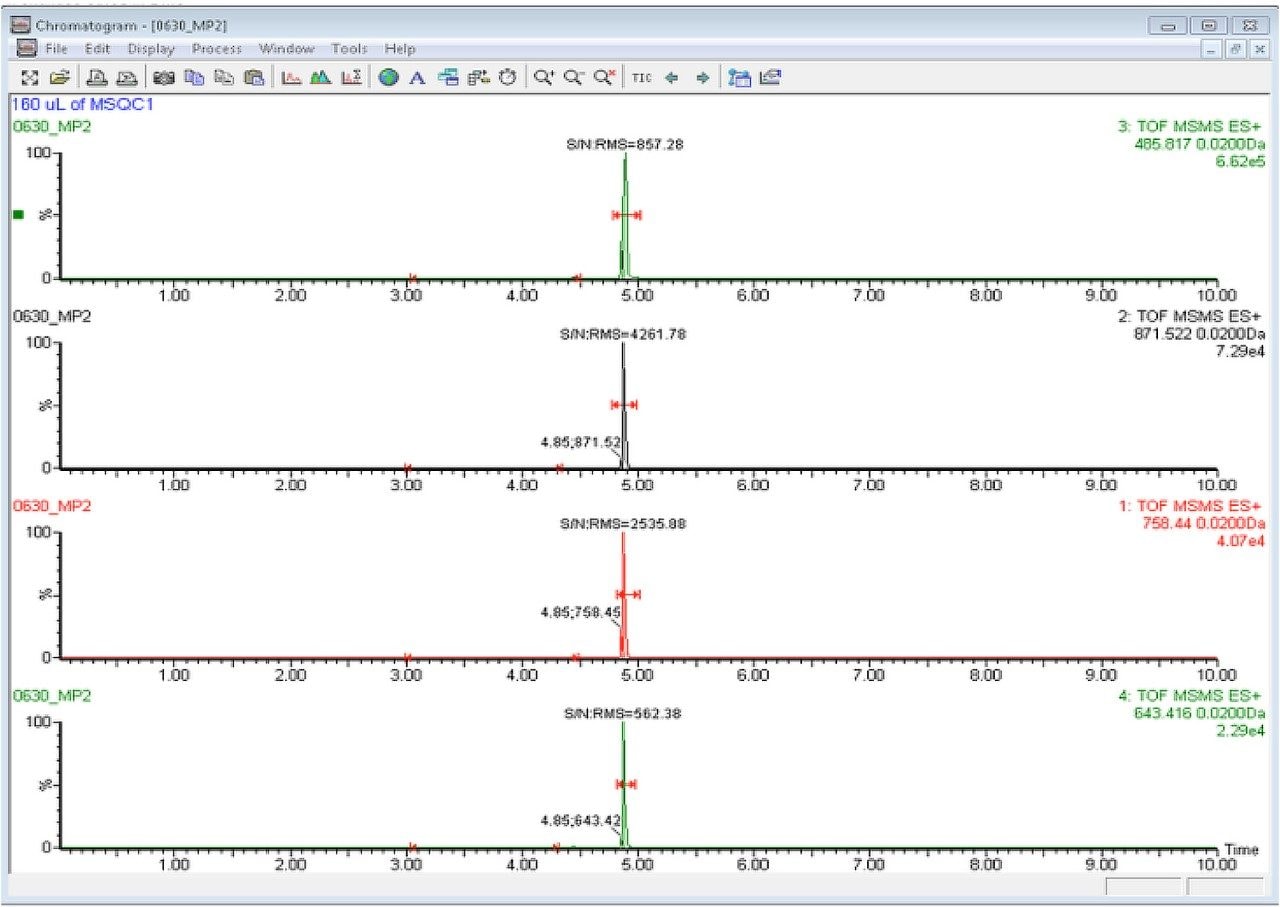
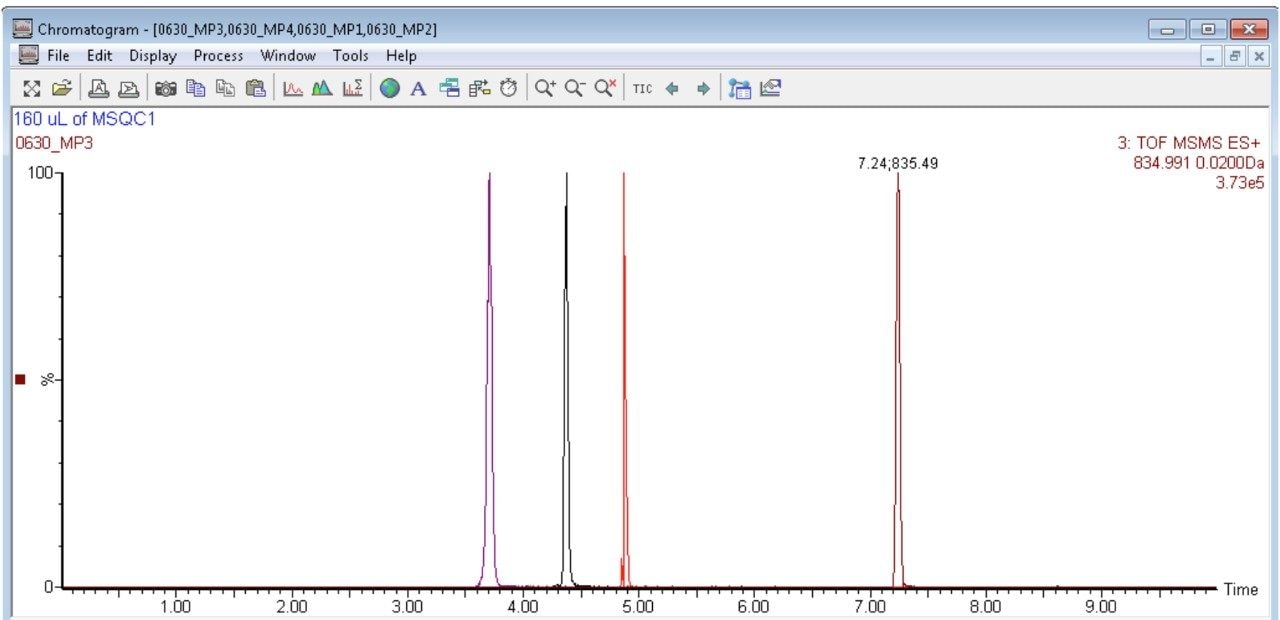
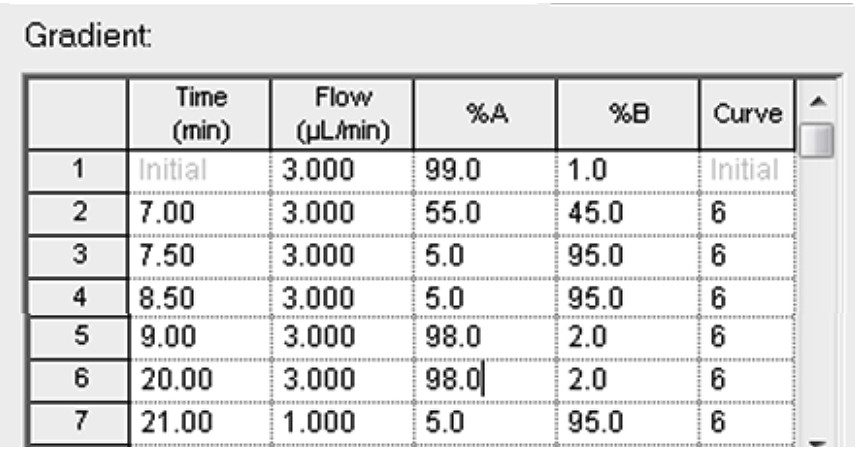
In this app brief, peptide quantitation using HRMS is demonstrated using ionKey/MS and Xevo G2-XS with Tof MRM mode for data acquisition (although the acquisition/data processing is amenable to all inlets available at Waters). Tof MRM, or target enhancement mode of acquisition, provides a selectivity and sensitivity boost for enhanced HRMS quantitation. Microfluidics such as ionKey also produce enhanced selectivity and sensitivity across small and large molecules, particularly with intact molecule characterization for both surrogate and intact quantitation.