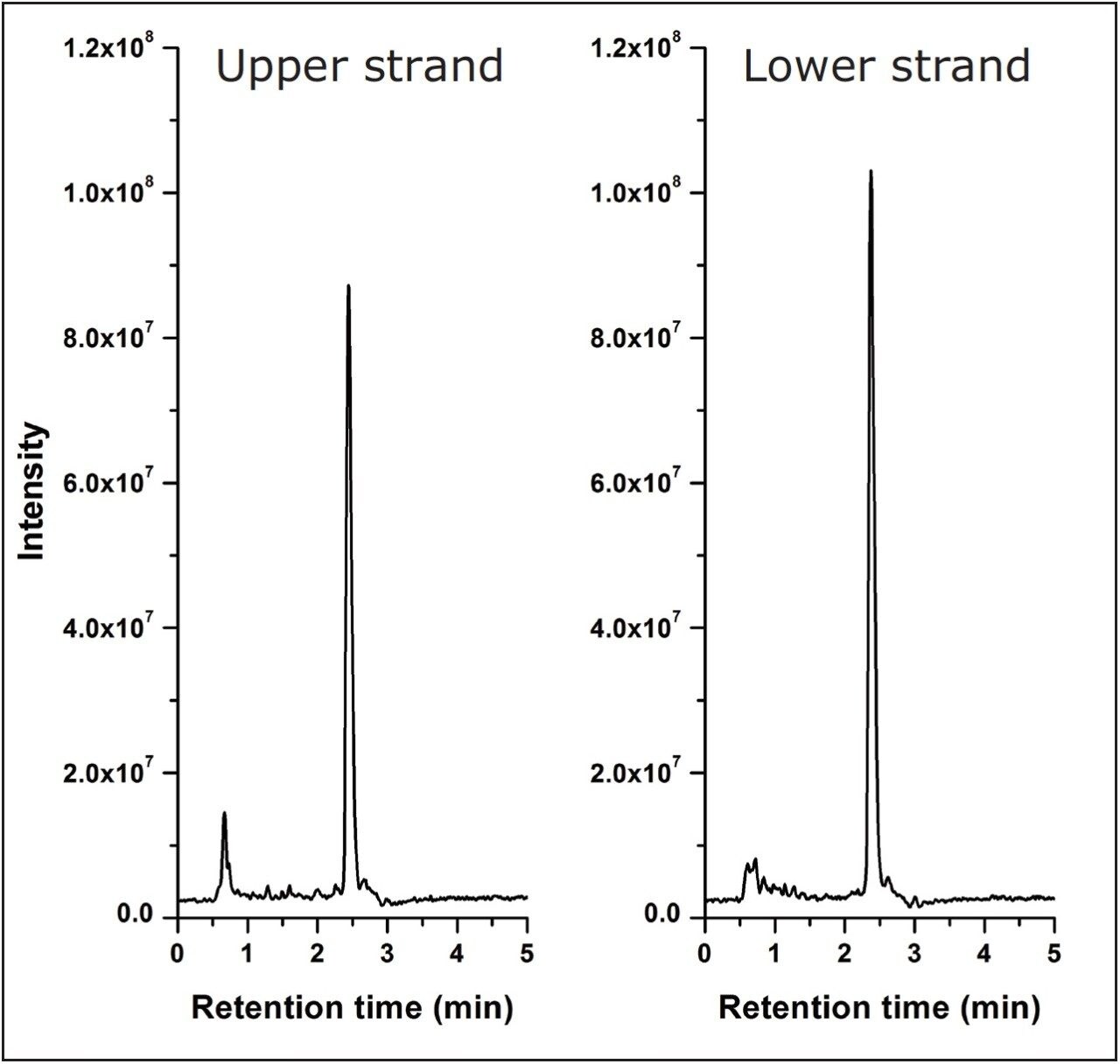
Research into therapeutic oligonucleotides has received steadily increasing attention from the pharmaceutical industry. This is due to potential applications using deoxyribonucleic acid (DNA) sense/antisense oligonucleotides and interfering ribonucleic acid- (RNAi) based therapies.1,2 The production of oligonucleotides with high yields via automated stepwise synthetic methods is well established. As part of the production process, purification and desalting steps are incorporated to remove byproducts of synthesis, such as failed sequences and production impurities. Characterization of purified synthetic products must be carried out prior to use in therapeutic applications to ensure product identity and purity. Ion Pairing Reversed Phase Liquid Chromatography (IP-RPLC) has become a prevalent technique in the analysis of synthetic oligonucleotides in part due to the selectivity offered by such techniques, as well as its ability to incorporate mass spectrometry-friendly reagents and buffers as first demonstrated by Apffel and colleagues.3,4
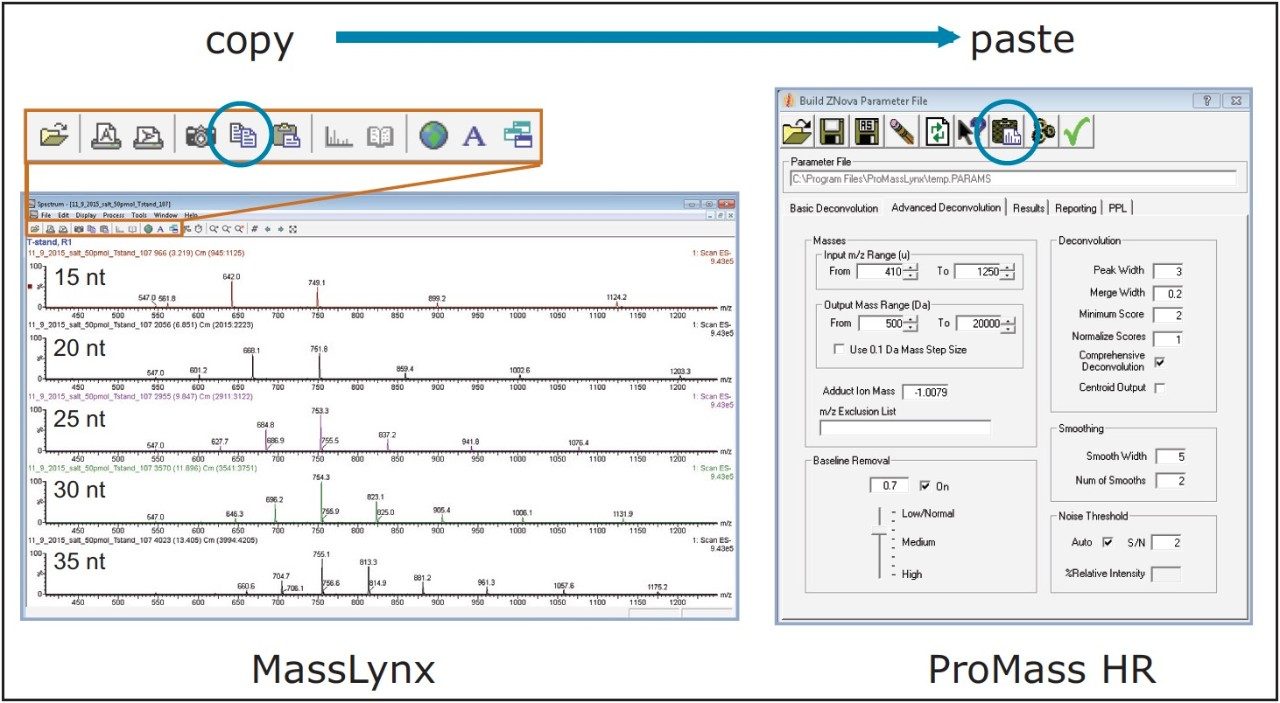
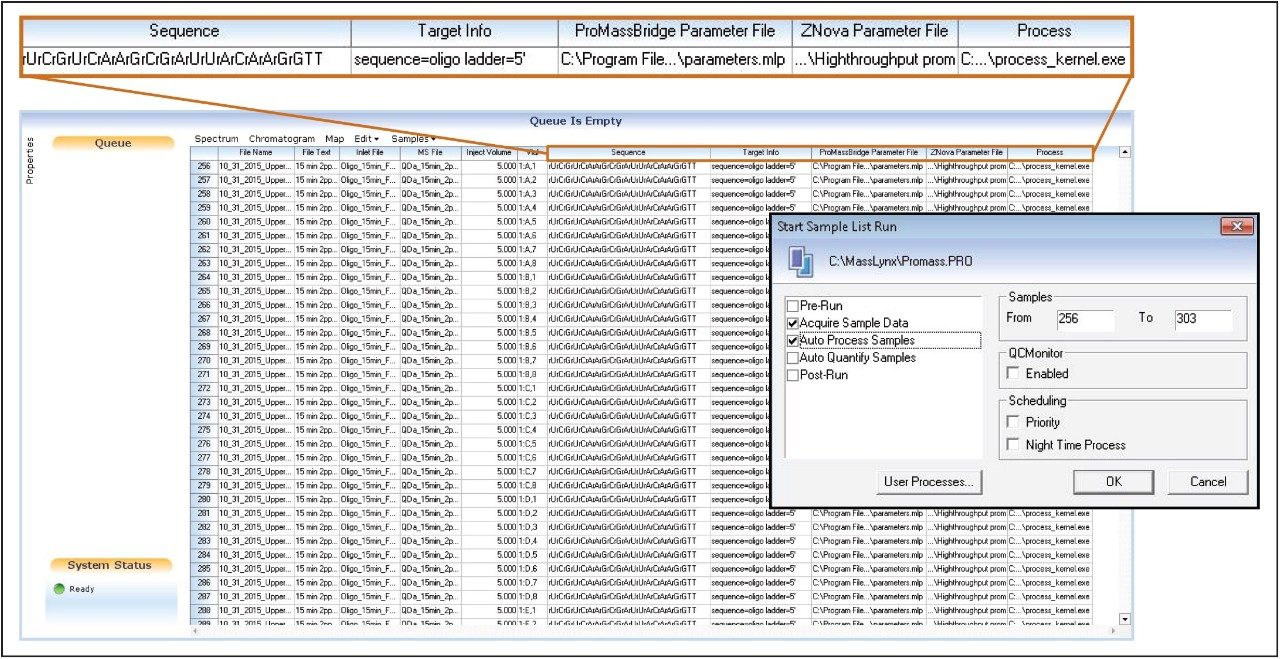
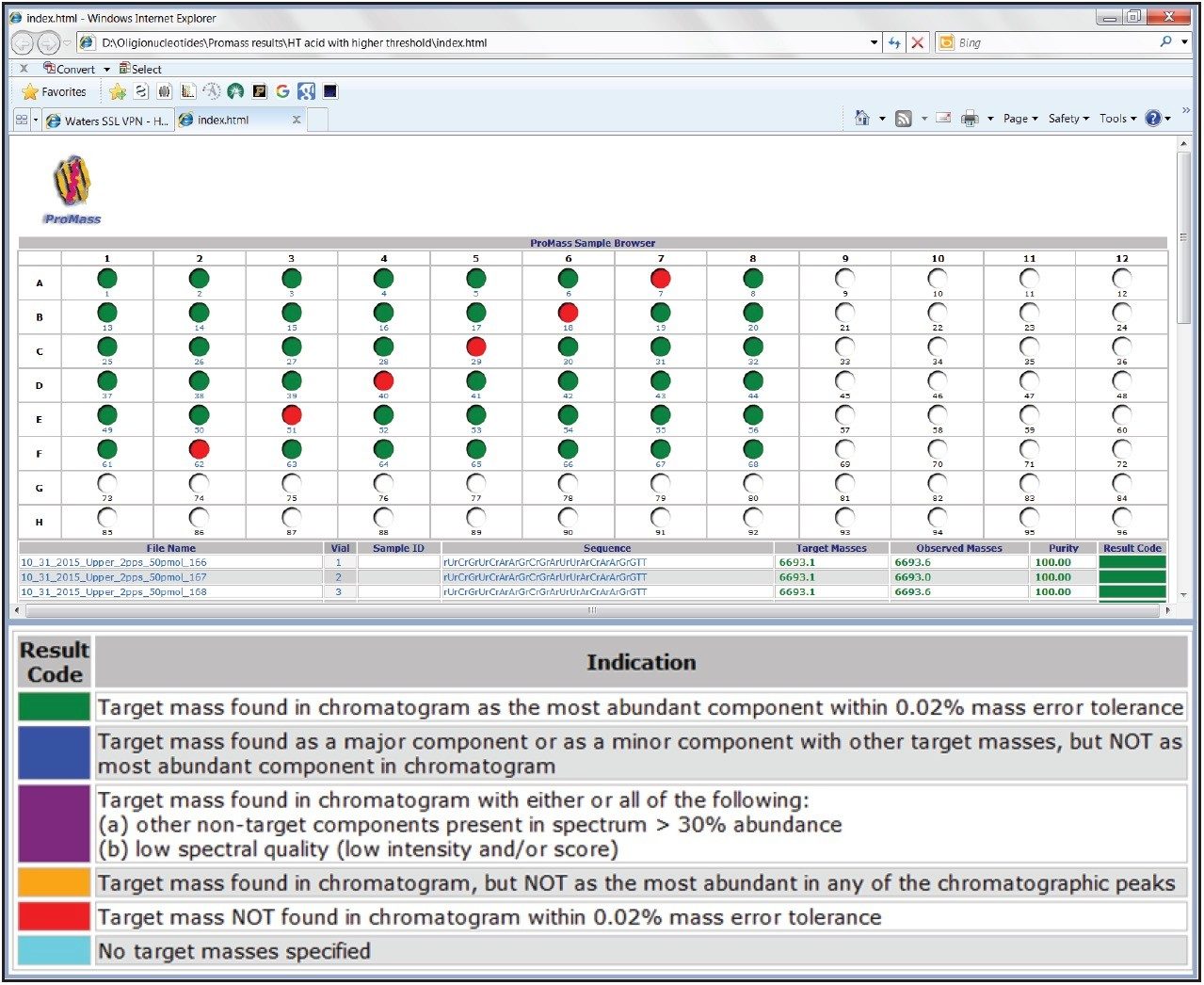
Mass information afforded by MS detection offers an efficient means of identifying challenging base modifications for improved productivity in synthetic therapeutic oligonucleotide workflows. Pharmaceutical companies engaged in oligonucleotide research are often investigating numerous potential biotherapeutic candidates, which can negatively impact productivity as the characterization process of synthetic oligonucleotides often requires manual processing of LC-MS data. Previous work demonstrated that incorporation of ProMass (Novatia, LLC) for MassLynx Software enables automated spectrum deconvolution and data analysis for high-throughput screening of UPLC-MS data generated on Waters MS instruments, such as the ACQUITY SQD Detector and SYNAPT.5 As one of the newest mass detectors from Waters, the ACQUITY QDa has been established as an efficient means for obtaining mass information within existing, optically-based LC workflows in the biopharmaceutical manufacturing environment when used as an orthogonal detection technique.6-8
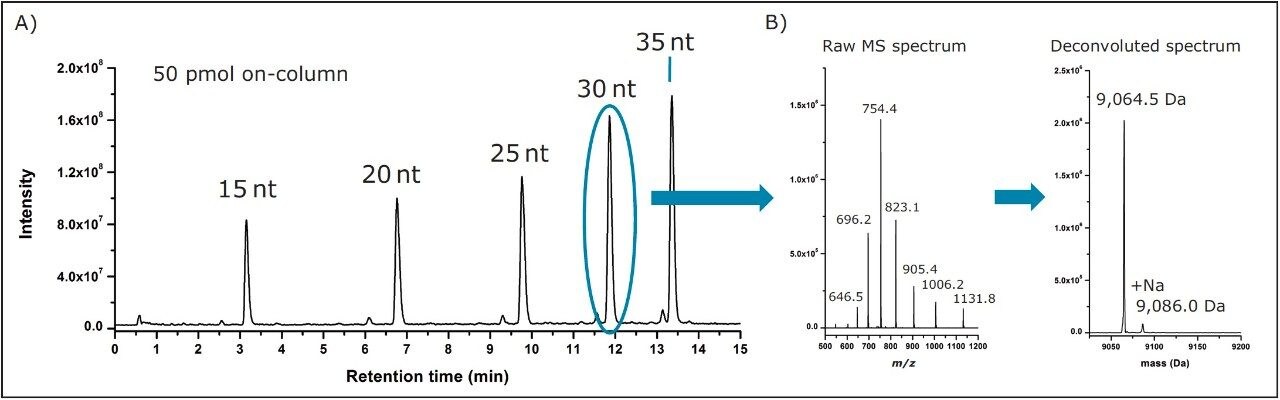
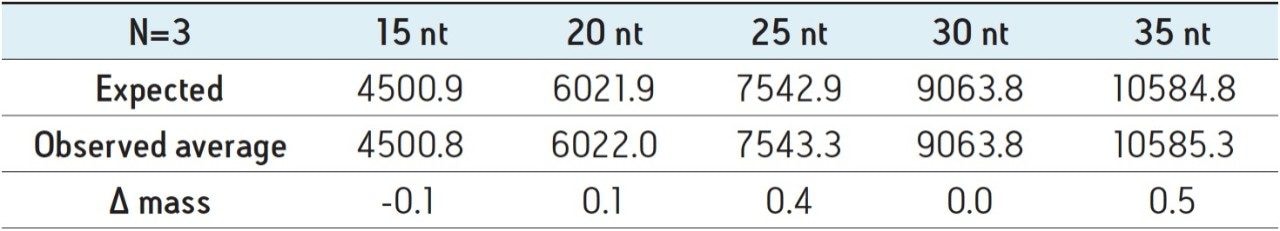
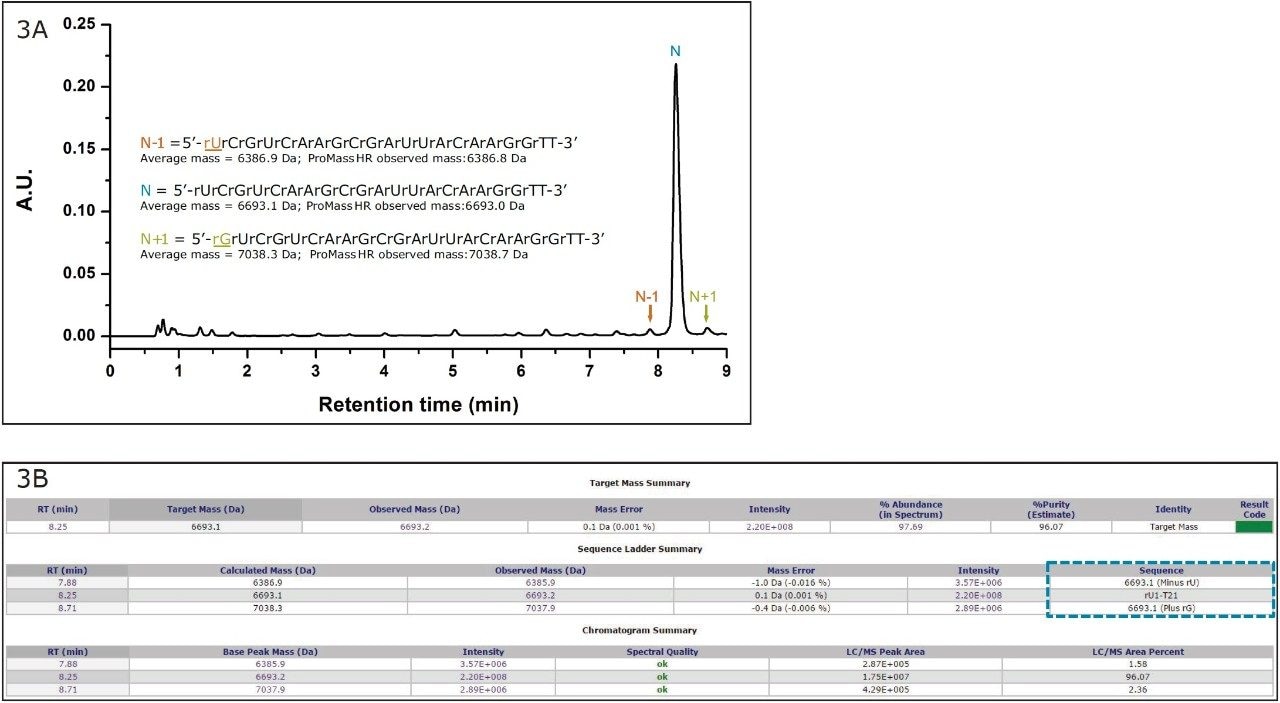
The objective of this application note is to demonstrate that the ACQUITY QDa Detector provides a simple and cost-effective solution in the assessment of identity and purity of synthetic oligonucleotides and that mass spectral data acquired can be readily processed with ProMass for MassLynx Software in an automated fashion.