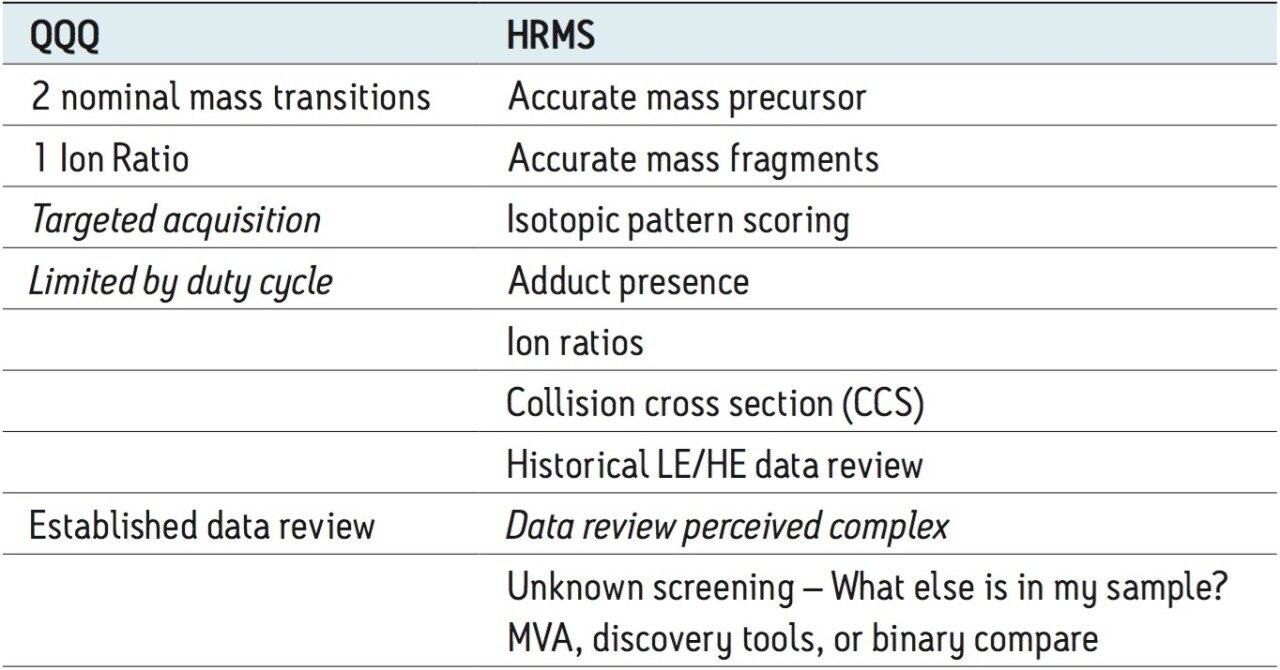
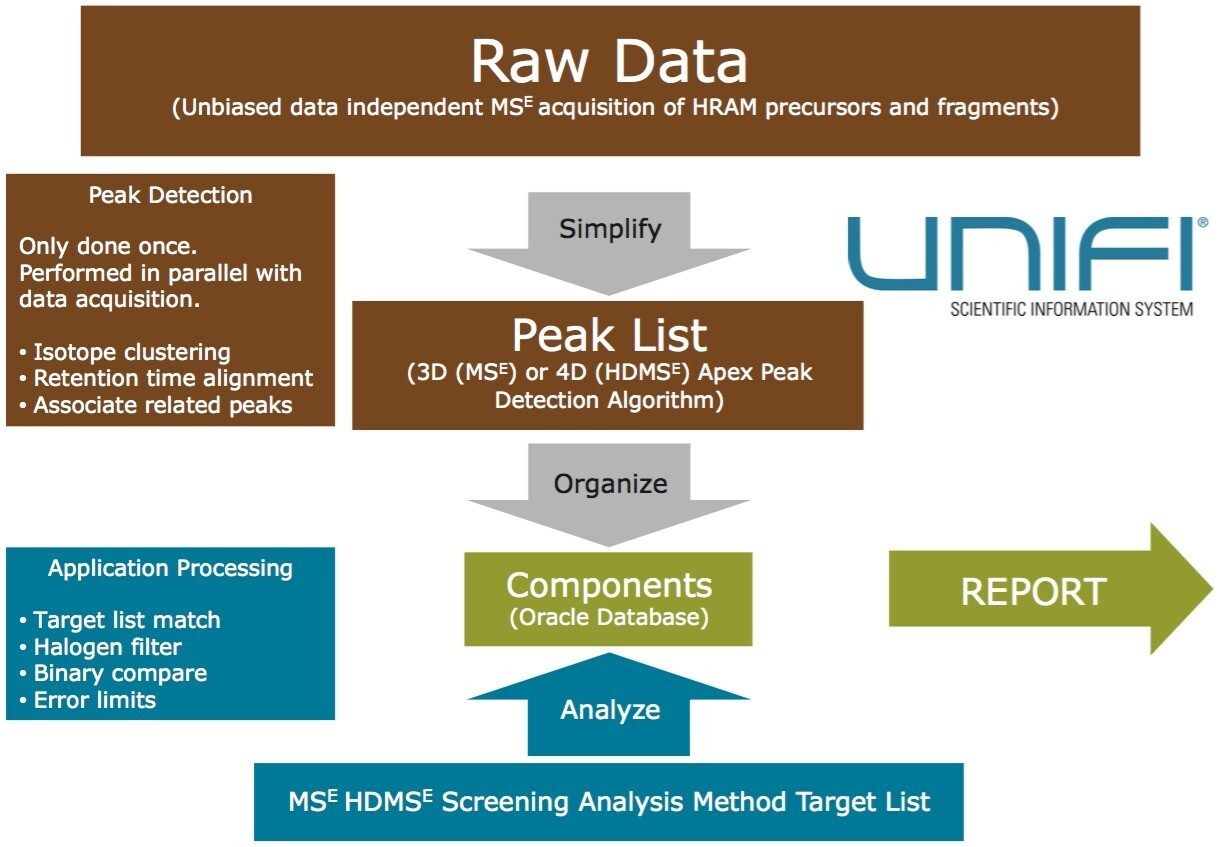
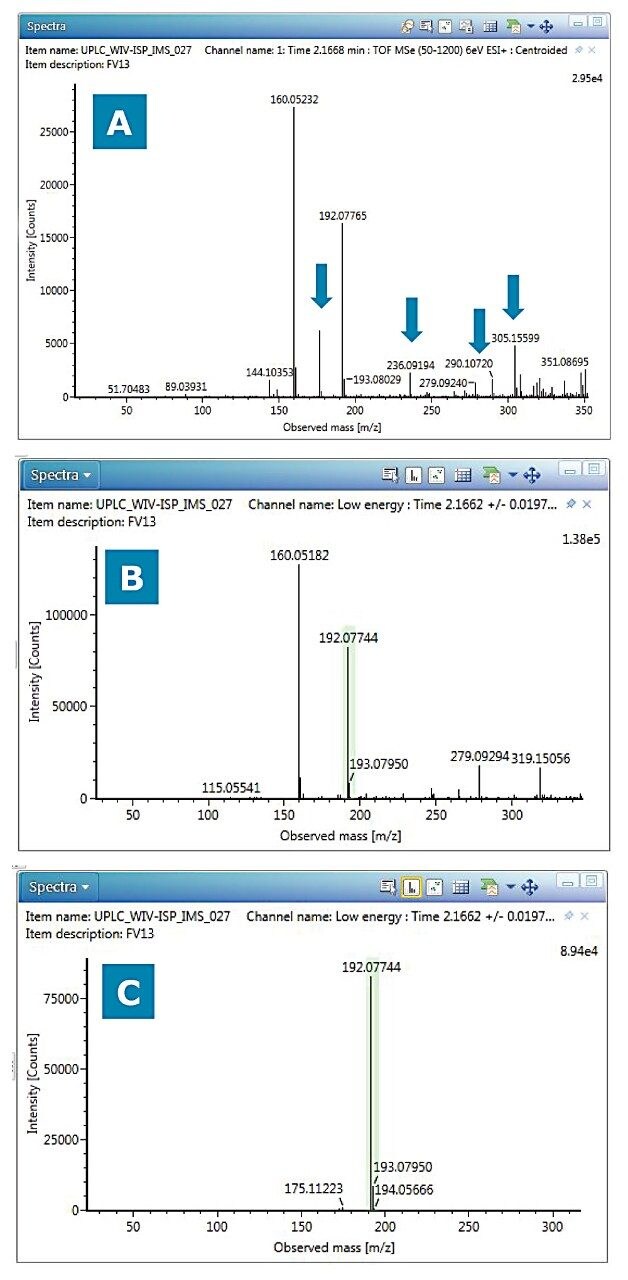
Data review
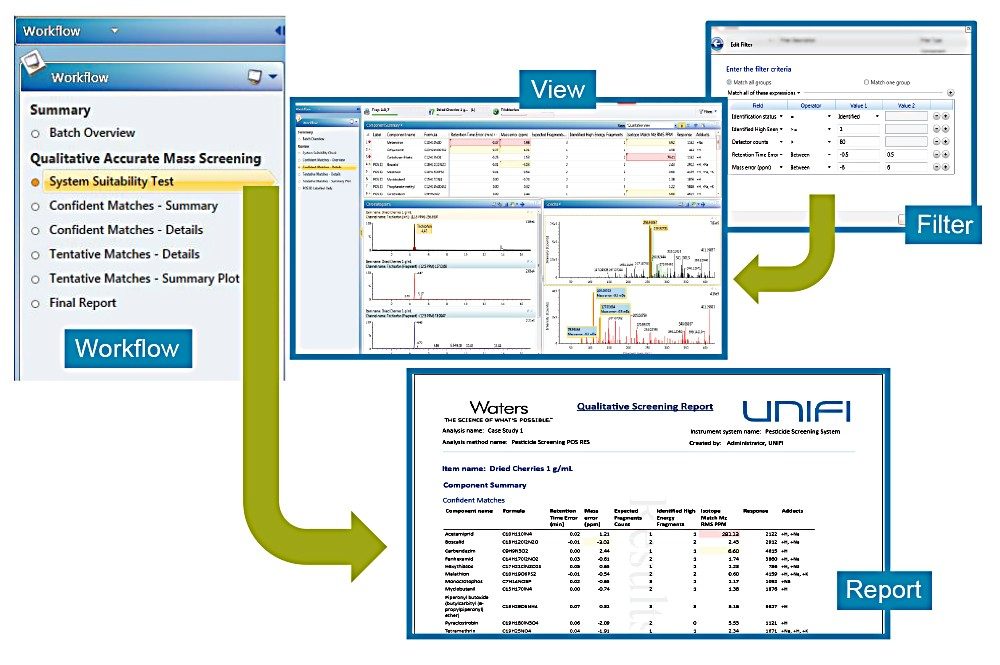
Interrogation of non-targeted, componentized MSE, or HDMSE, data in UNIFI is performed using filters, workflows, and views.
A filter is a question or a means to interrogate the componentized data generated in UNIFI. For example, “Show me the components identified with mass accuracy (±5 ppm), retention time (±0.2 mins), and the presence of a high energy accurate mass fragment ion.” A second example could come from an unknown screening perspective such as “Show me components with a high probability of containing a halogen atom.” Another example for interrogation of unknown compounds of interest would be “Show me all components with a common accurate mass fragment of 180.0634 Da.”
A view is the combination of plots, chromatograms, spectra, tables, and columns that are displayed together on the screen. The view visually provides all the information required to answer the question in a filter.
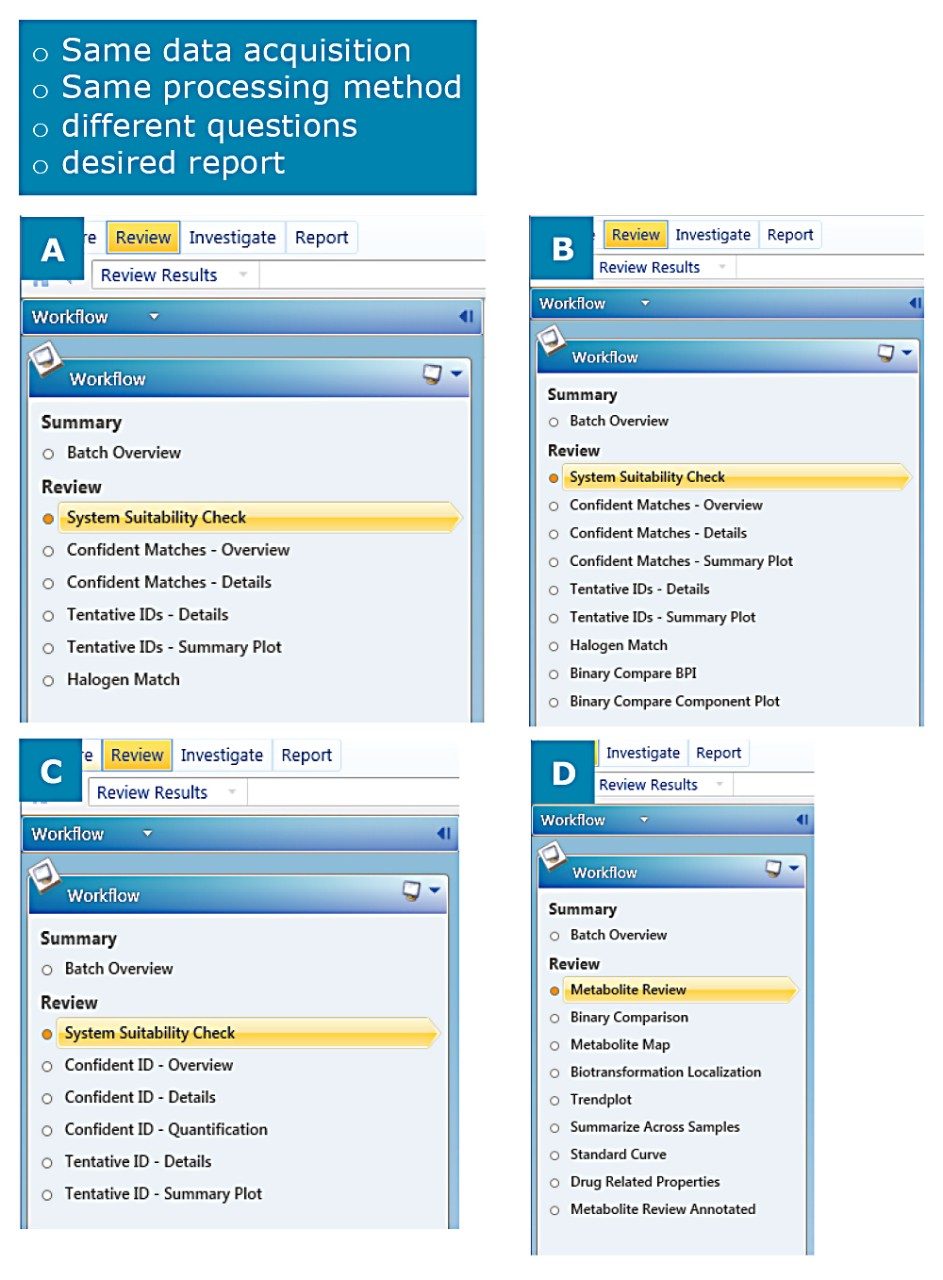
A workflow step is simply a saved view with a filter applied. A combination of these steps creates the workflow, which is designed to answer a series of targeted and/or unknown screening questions for each injection within an analysis.
The workflow allows a supervisor, for example, to determine what information to extract from a non-targeted acquisition and customize how the review process is implemented. This ensures that the time from injection to report is minimized and that all users review data in a consistent and concise manner.
Figure 4 illustrates the interaction of filters, workflows and views designed to facilitate getting a user from injection to report as fast as possible.