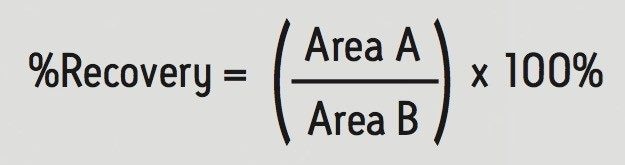
Sample preparation is an important consideration for any bioanalytical LC-MS/MS method designed for forensic toxicology. Waters has developed a novel sample preparation sorbent, Oasis PRiME, which is designed to have some key advantages over traditional SPE sorbents. These include the ability to eliminate sorbent preconditioning and equilibration, allowing a more rapid workflow compared to traditional SPE products, and the ability to remove more interferences, resulting in a cleaner extracts and reducing the risk of short column lifetimes or MS source fouling.
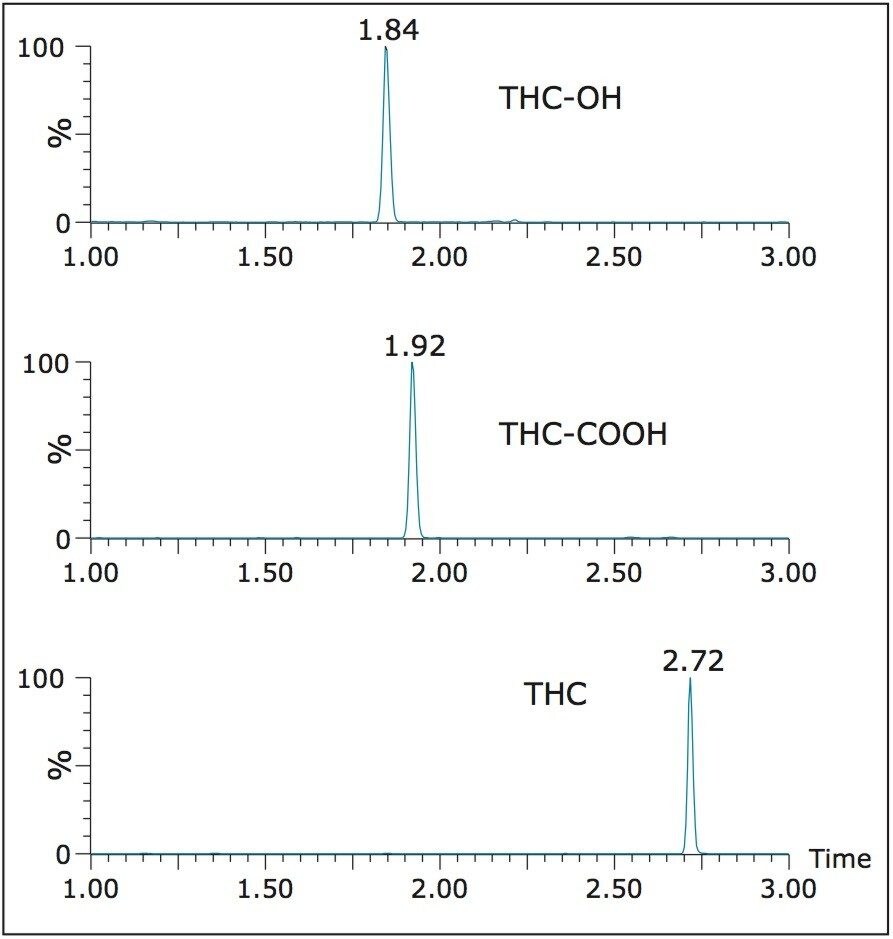
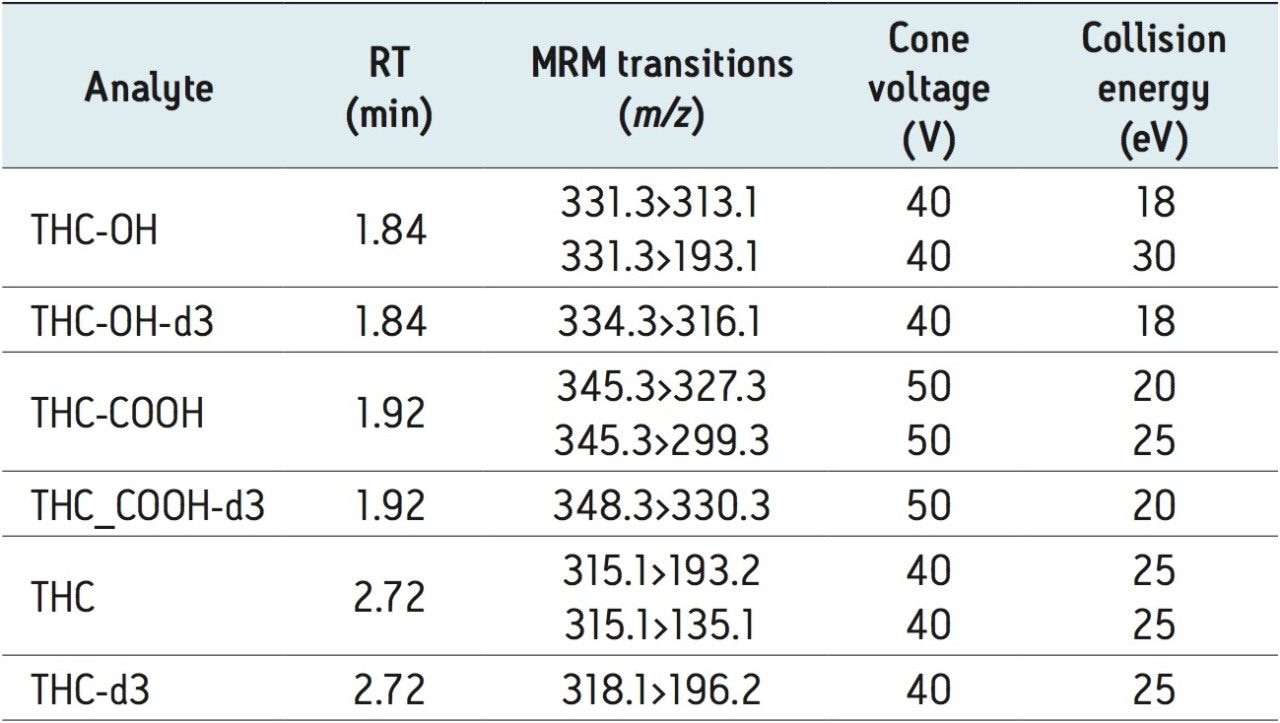
This application note details the extraction and UPLC-MS/MS analysis of Δ-9-tetrahydrocannabinol (THC) and its metabolites, 11-hydroxy- Δ-9-THC (THC-OH) and 11-nor-9-Carboxy-Δ-9-THC (THC-COOH) from urine using Oasis PRiME μElution Plates. Δ-9-tetrahydrocannabinol (THC) is the main psychoactive element present in the plant Cannabis sativa. Quantitative analysis of these compounds in urine is an indicator of cannabis consumption, with high levels indicating recent and/or chronic use.
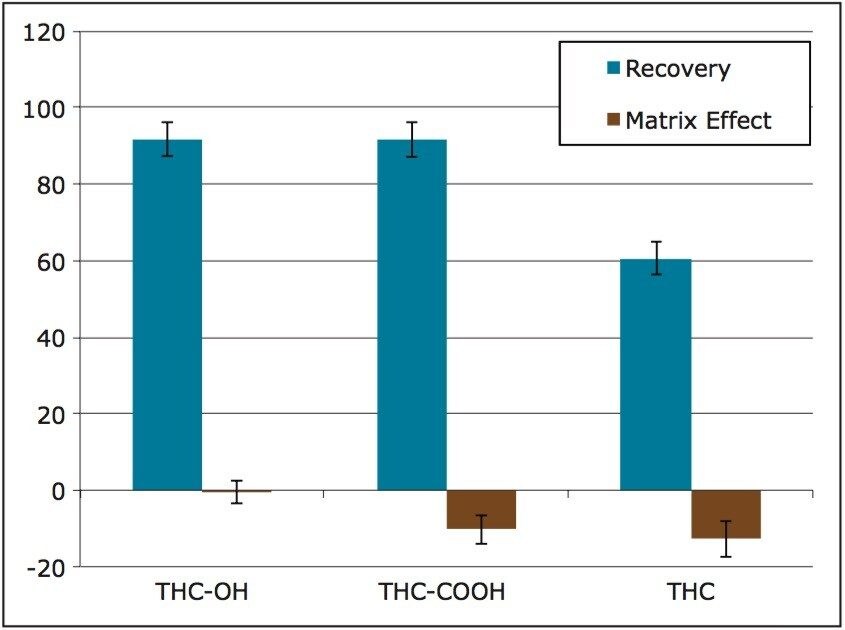
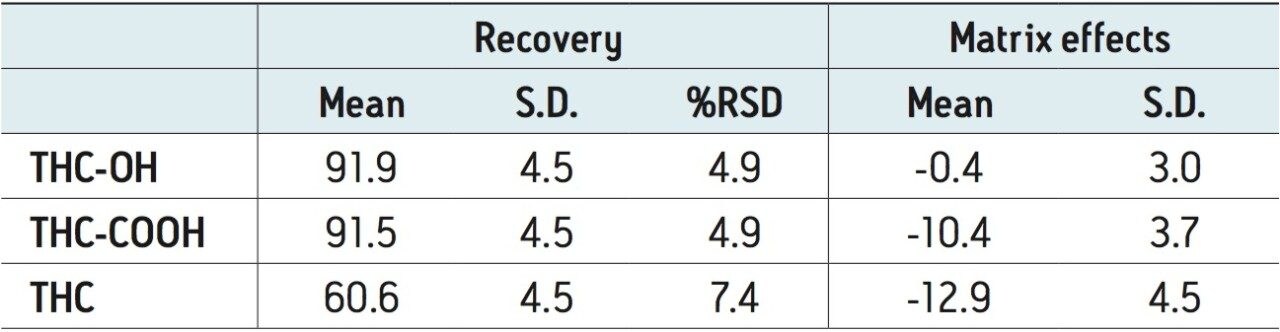
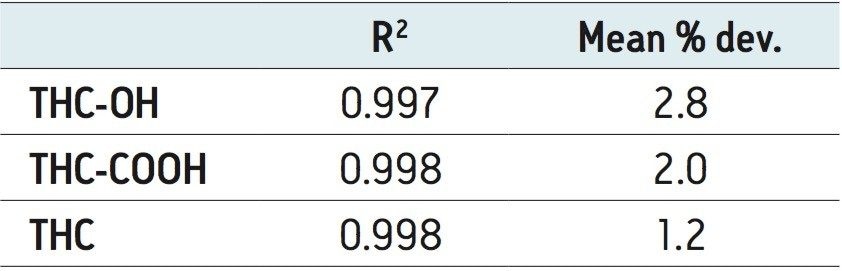
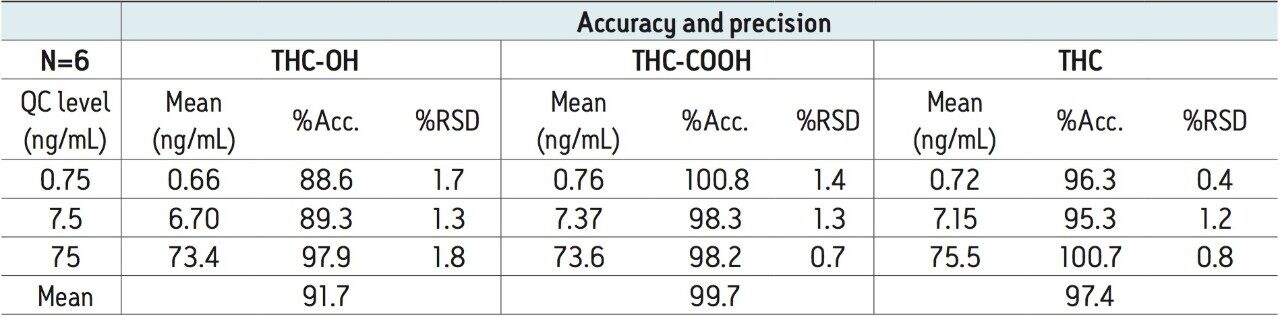
The use of Oasis PRiME resulted in consistent and highly reproducible recoveries of all compounds with minimal matrix effects. The μElution format allowed for the concentration of the sample on the SPE column, eliminating the need to evaporate and reconstitute the sample, minimizing the risk of analyte loss due to nonspecific binding and streamlining the laboratory workflow. This resulted in a method that was linear, accurate and precise for all analytes, with limits of quantification of 0.1 ng/mL for THC and its metabolites.