It is well known that thermal, viscosity, and eluent mismatches can cause strange peak profiles.3-7 As a best practice, dissolving and injecting a sample prepared in the exact same composition as the mobile phase (or initial mobile phase conditions of a gradient) is recommended. Unfortunately, dissolving and injecting a sample prepared in a supercritical fluid is difficult and very rarely done. Alternatively, an analyst could prepare a sample in a small extraction or dissolution vessel and use the compressed mobile phase to instantly dissolve and carry the sample to a column inlet. Extraction vessels have been used successfully on supercritical fluid extraction (SFE) and preparative SFC systems; however, it requires the sample be dissolved instantly and completely by the effluent, making broad implementation difficult. Those factors make choosing the right sample solvent or diluent paramount to achieving optimal peak profiles for routine analytical injections. It should also be noted that the Partial Loop with Needle Overfill (PLNO) method of injection used by ACQUITY UPC2 systems will also inject a small amount of ‘weak needle wash’, which is used to transfer the sample liquid from a vial to the injector loop. As the name implies, a weak eluent is necessary to prevent disruption of the injected sample band. The experiments performed in this application only address the effects of injection solvent and analyte polarity in chiral UPC2 separations. Sample loading was not a consideration and should be examined on a case-by-case basis.
Sample diluent induced peak distortion is due to a disruption of the equilibrium between mobile and stationary phases. When a sample is injected into a mobile phase stream of a different composition, e.g. a weaker or stronger elution solvent, the local mobile phase composition alters the peak elution profile. The distortion occurs briefly at the inlet of a column bed. In the case of using a sample diluent weaker than the mobile phase, peak sharpening or focusing will occur. When the sample diluent has a stronger elution strength than the mobile phase, peak broadening will occur. We have found that distorted peak profiles caused by the strong solvent effect (injecting a sample diluent having a stronger eluent than the mobile phase) are very repeatable. It should be noted that there are destructive solvents which should never be used as injection solvents or in mobile phases on Trefoil phases: tetrahydrofuran (THF), dimethylsulfoxide (DMSO), dichloromethane (DCM), and other halogenated solvents.8
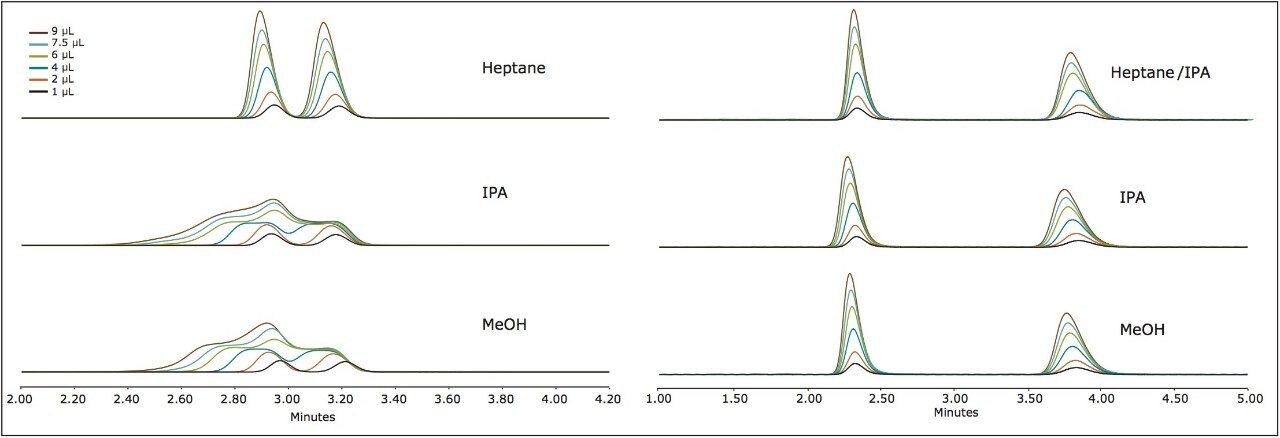
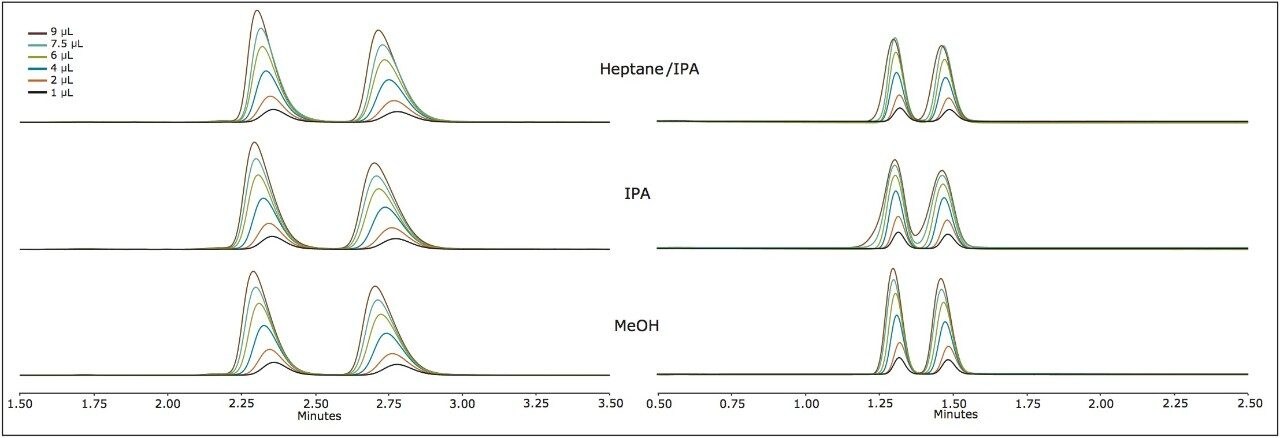
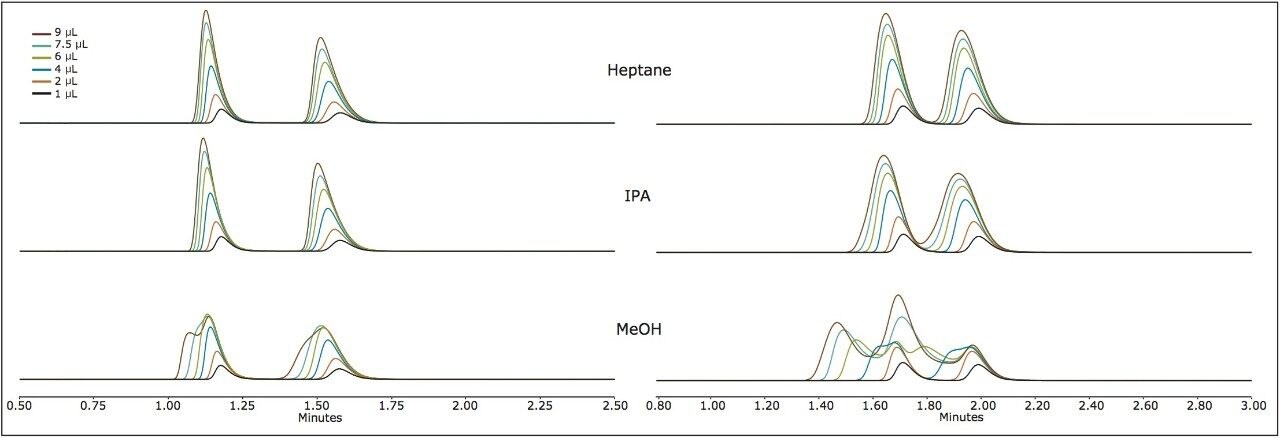
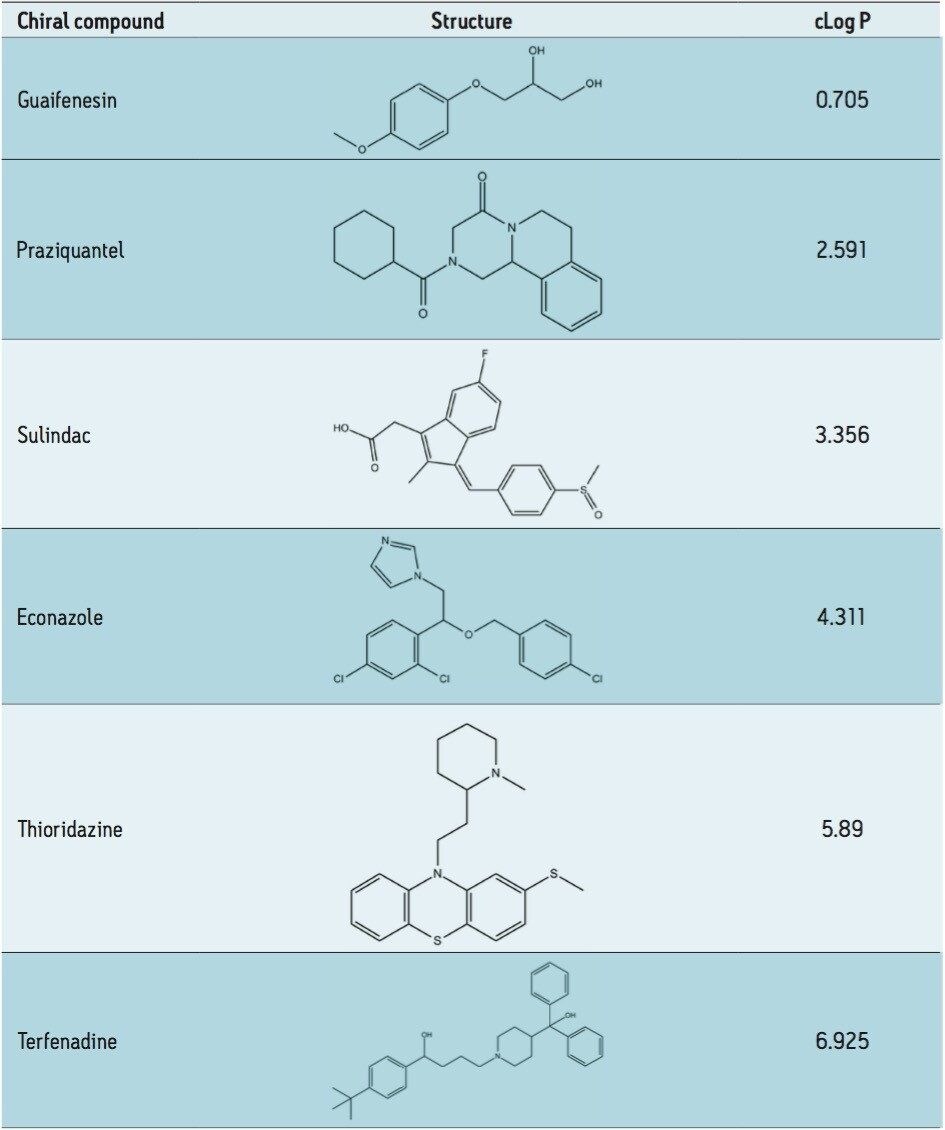
Suitable chiral separation conditions were found for each of the chiral analytes using the Trefoil AMY1 column, with the exception of terfenadine, which required the Trefoil CEL1 chemistry. A single co-solvent was employed and used for both isocratic and gradient elutions. The co-solvent percentage was adjusted for each compound to achieve retention times of less than five minutes for each analyte. The injection volume was increased for each sample, but never to a point of producing a mass overloaded peak profile. Mass overloading is not a concern in these experiments. For example, if we look at the peak profiles of the same injection volume across different sample diluents, the peak profiles can be very different, e.g. guaifenesin, or similar, e.g. praziquantel, but none of the results were characteristic of mass overloading. Furthermore, all the samples were prepared in dilute solutions, around 0.4 g/L, and injection volumes were less than 2.5% of the column hold-up volume. The initial observations of peak distortion show broadening of the peak, particularly near the peak apex. As the injection volume increases, peak distortion increases and has the appearance of a shouldering second peak. In the case where there are two closely eluting peaks, the profiles can be distorted so much that the two peaks can appear to be one or even three peaks. This can be a significant problem for chiral and preparative separations. Chiral separations limit the analyst’s ability to easily distinguish between the distorted peak profiles, as UV and MS signals are identical for a pair of enantiomers. In Figure 1, two contrasting results are seen between guiafenesin and praziquantel. The former peaks are highly distorted with the use of IPA or methanol as the injection solvent, while the latter is nearly unaffected by the choice of sample diluent. The relatively high retention (33% co-solvent was used for elution) for praziquantel and the mid-range cLog P value work together to mitigate the effect of sample diluent.