The analysis of natural and synthetic opioid drugs continues to be an important aspect of forensic toxicology. A substantial percentage of arrests and/or deaths are attributed to the misuse or abuse of narcotic pain relievers such as oxycodone and hydrocodone, as well as the illegal opiate, heroin. Forensic laboratories often need to analyze whole blood specimens for the presence of different drugs to determine the precise cause of death, in cases of driving under the influence of drugs, or other criminal or research purposes. In the past, opioid analyses were typically conducted by GC/MS after first subjecting the samples to acid or enzymatic hydrolysis to liberate glucuronide metabolites.1 This step of using enzymatic hydrolysis to convert glucuronide metabolites to their free form adds time and expense to analysis, and complete and consistent hydrolysis is not always assured.2 With the advent of modern UPLC-MS/MS techniques, glucuronide metabolites can now be analyzed directly.3-6
Many sample preparation strategies have been used for whole blood analysis, including liquid-liquid extraction (LLE) and solid phase extraction (SPE). One of the simplest involves cell lysis followed by protein precipitation. The method presented in this study describes a rapid and straightforward sample preparation strategy using Ostro Sample Preparation Plates whereby whole blood samples can be pre-treated to lyse the cells, precipitated with acetonitrile, and eluted using a simple 96-well format. All sample pre-treatment is conducted within the wells of the Ostro Plate, without the need for centrifugation or sample transfer from individual tubes.
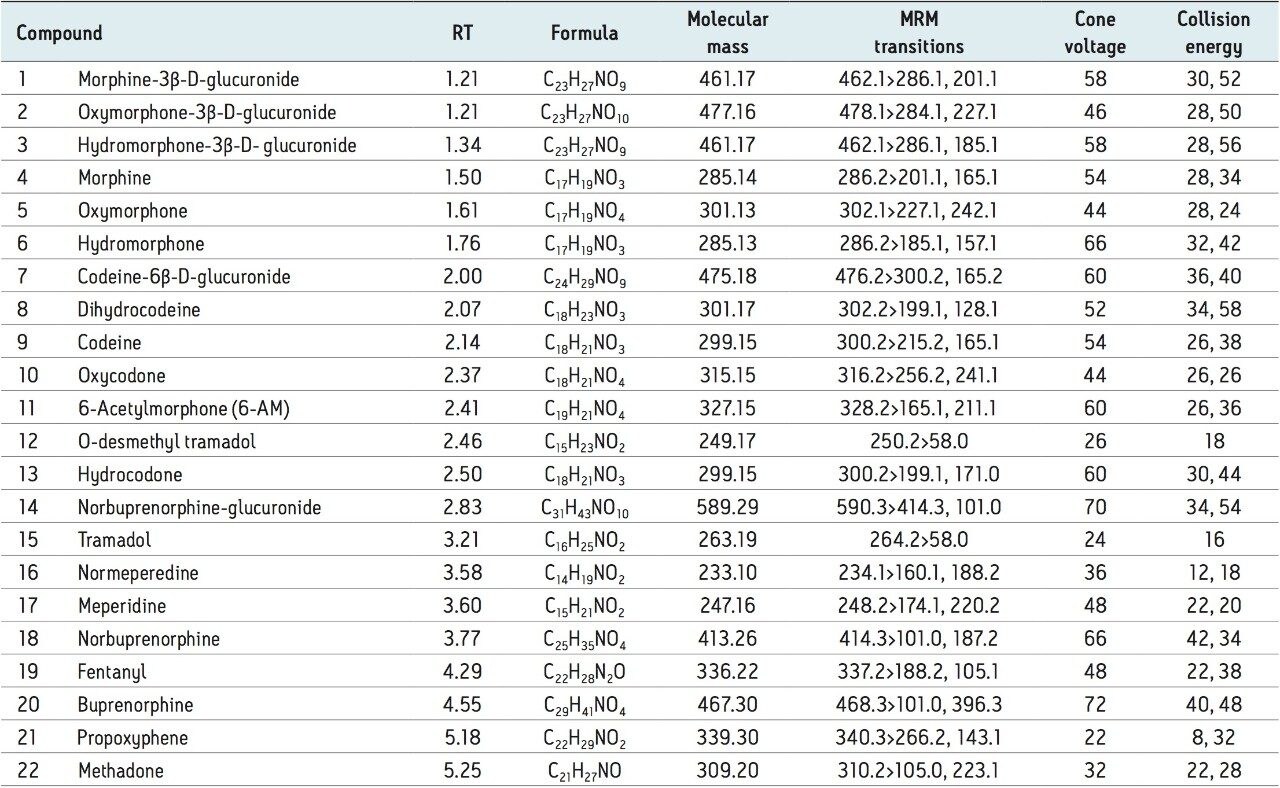
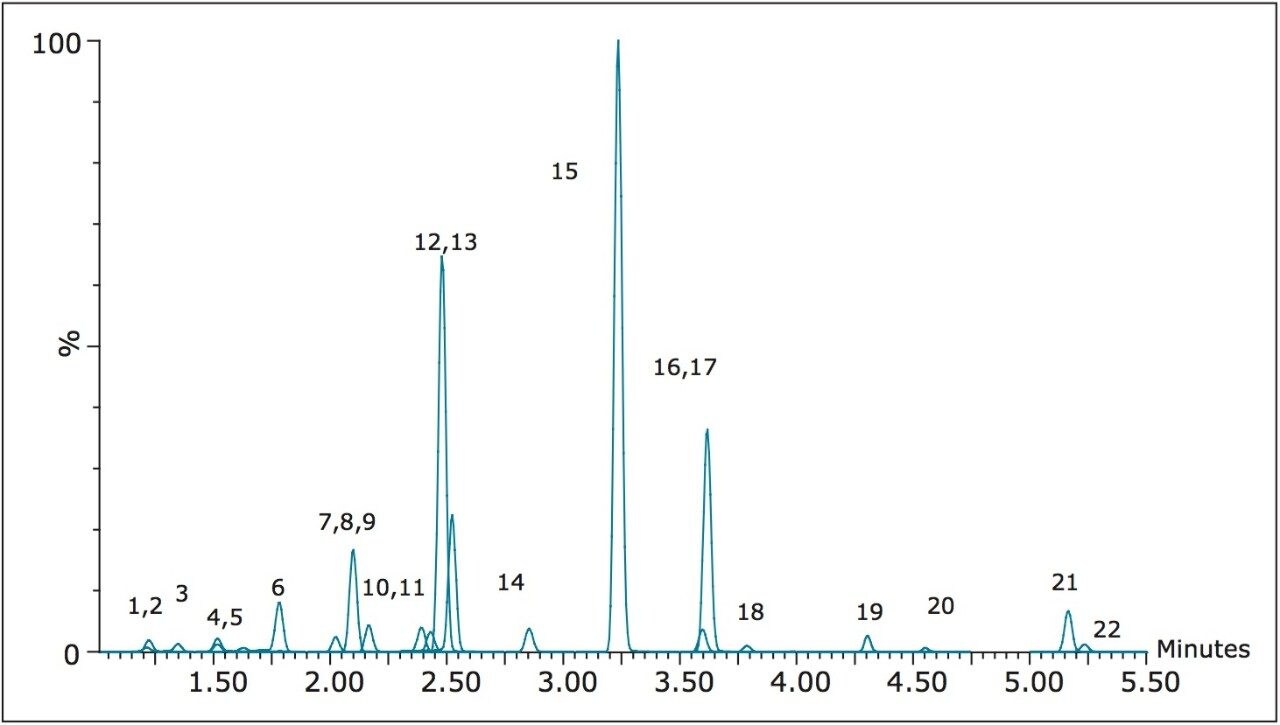
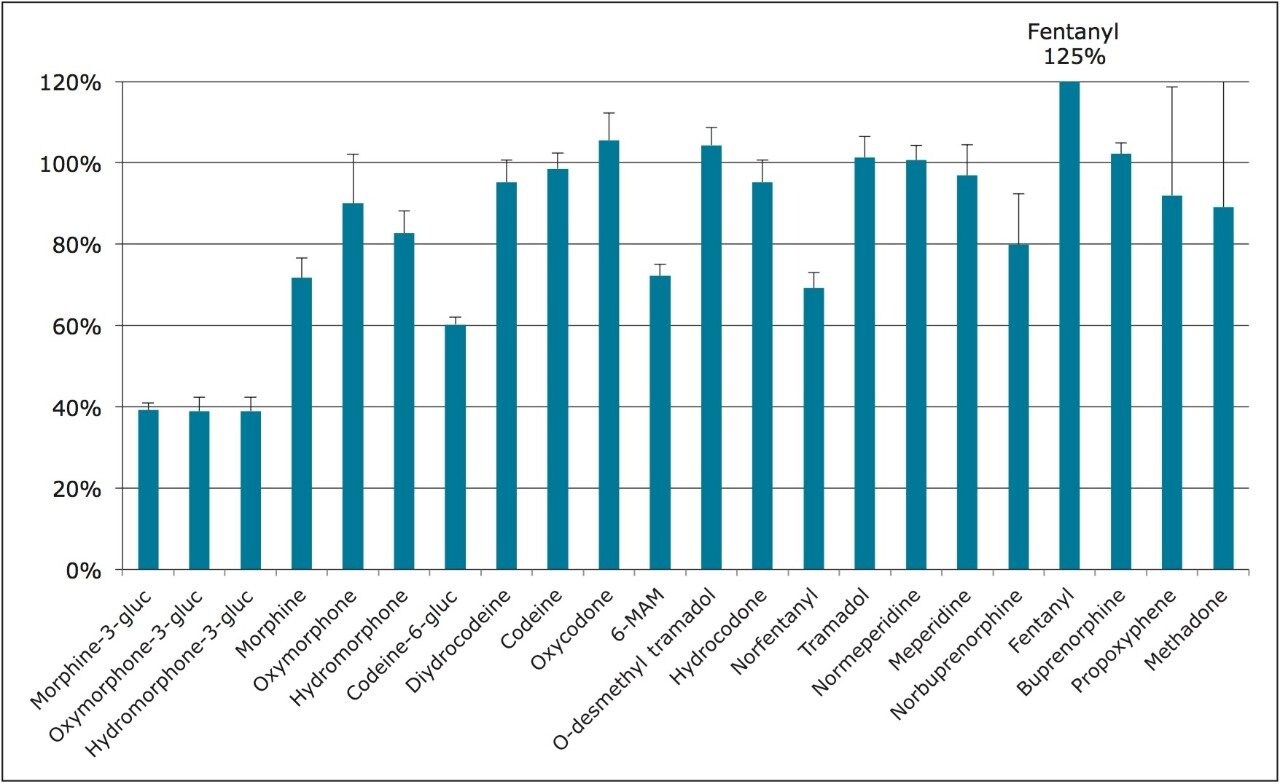
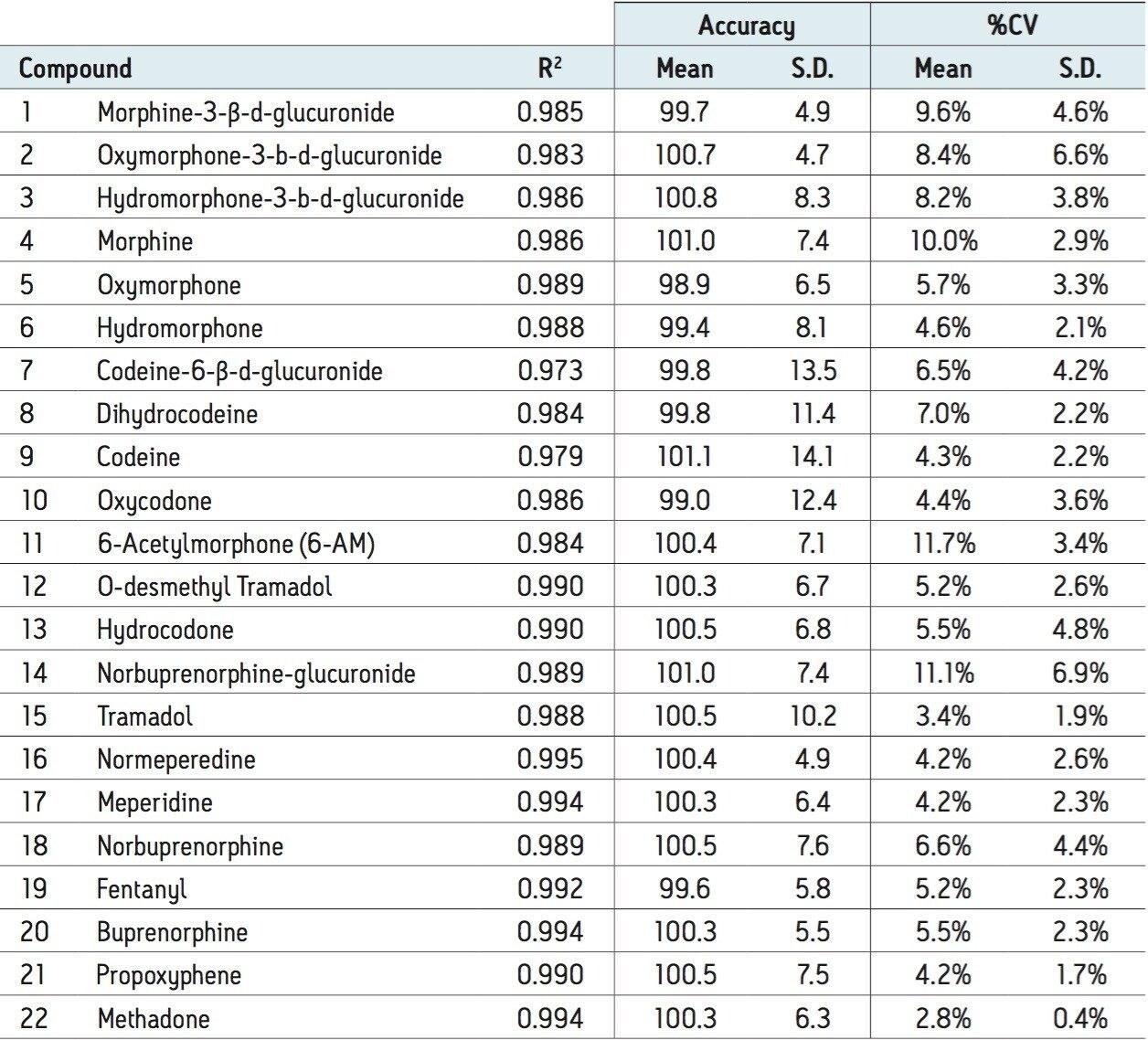
Following sample preparation, 22 opioid drugs and metabolites are subsequently analyzed by UPLC-MS/MS. Glucuronide metabolites are directly analyzed, eliminating the need for enzymatic or chemical hydrolysis. Calibration curves are linear with appropriate limits of detection easily reached.