Three replicates of each E.coli sample, differentially spiked with bovine serum albumin (BSA), alcohol dehydrogenase (ADH), enolase, and glycogen phosphorylase B were analyzed. The injected on-column amounts for the spike protein in the first sample (Mixture 1) were 500 attomoles each and 4000, 500, 1000, and 250 attomoles for the second sample (Mixture 2), respectively. The nominal expected ratios (Mixture 2:Mixture 1) were therefore 8:1, 1:1, 2:1, and 1:2. The peptides were separated and analyzed using a nanoACQUITY UPLC System coupled with a SYNAPT G2-S, operating at a mass resolution of > 20k FWHM. The data were acquired in LC-MSE mode, which is an unbiased TOF acquisition method in which the mass spectrometer switches between low and elevated energy on alternate scans. Post acquisition processing software correlates precursor and product ions by means of chromatographic retention times. Further correlation processing occurs during database searching the data that is based on the physicochemical characteristics of peptides when they undergo collision induced fragmentation. Searches and quantification were conducted with ProteinLynx Global SERVER v.2.5.1 using a species specific database where sequence information of the spiked proteins was appended.
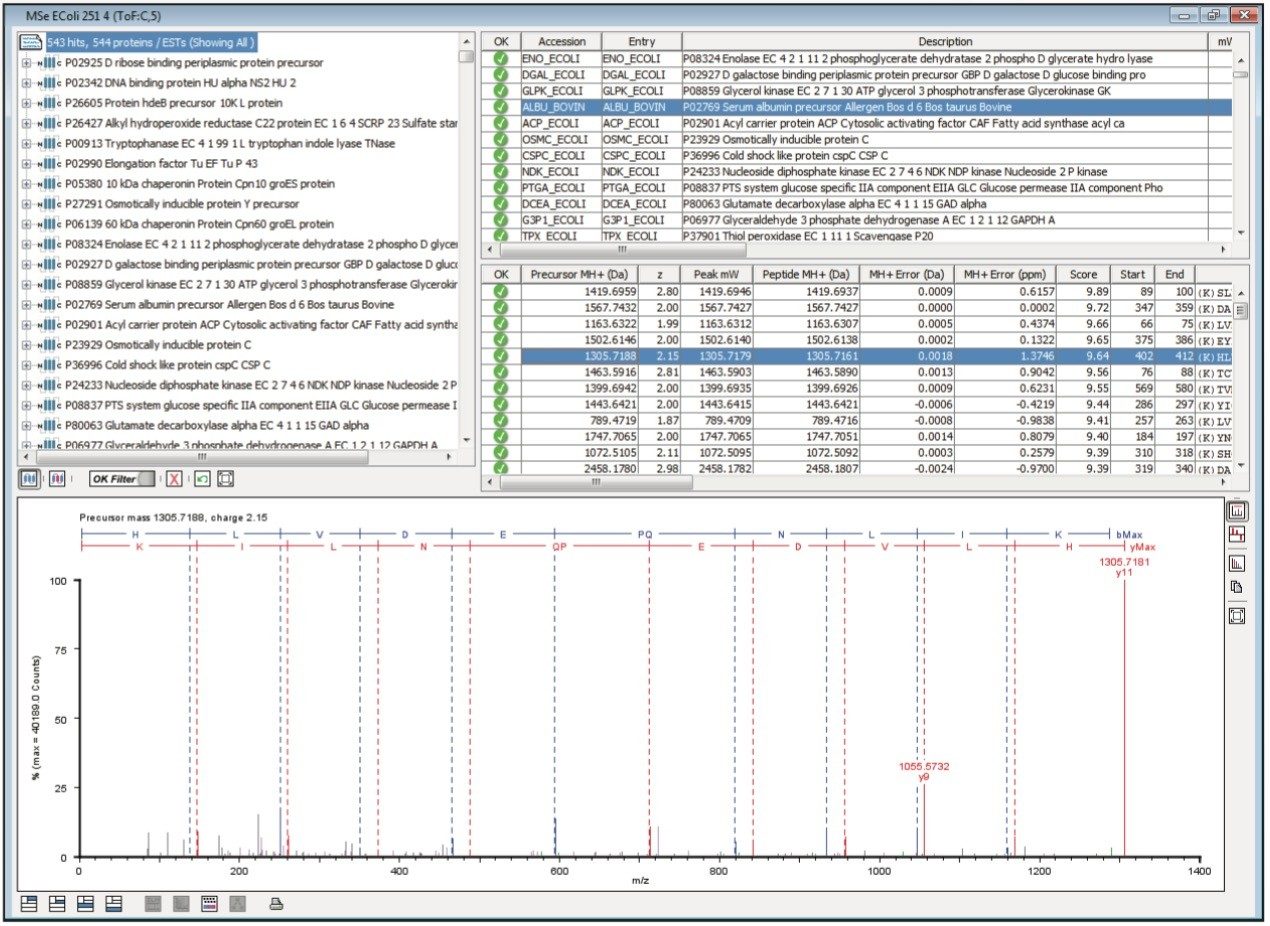
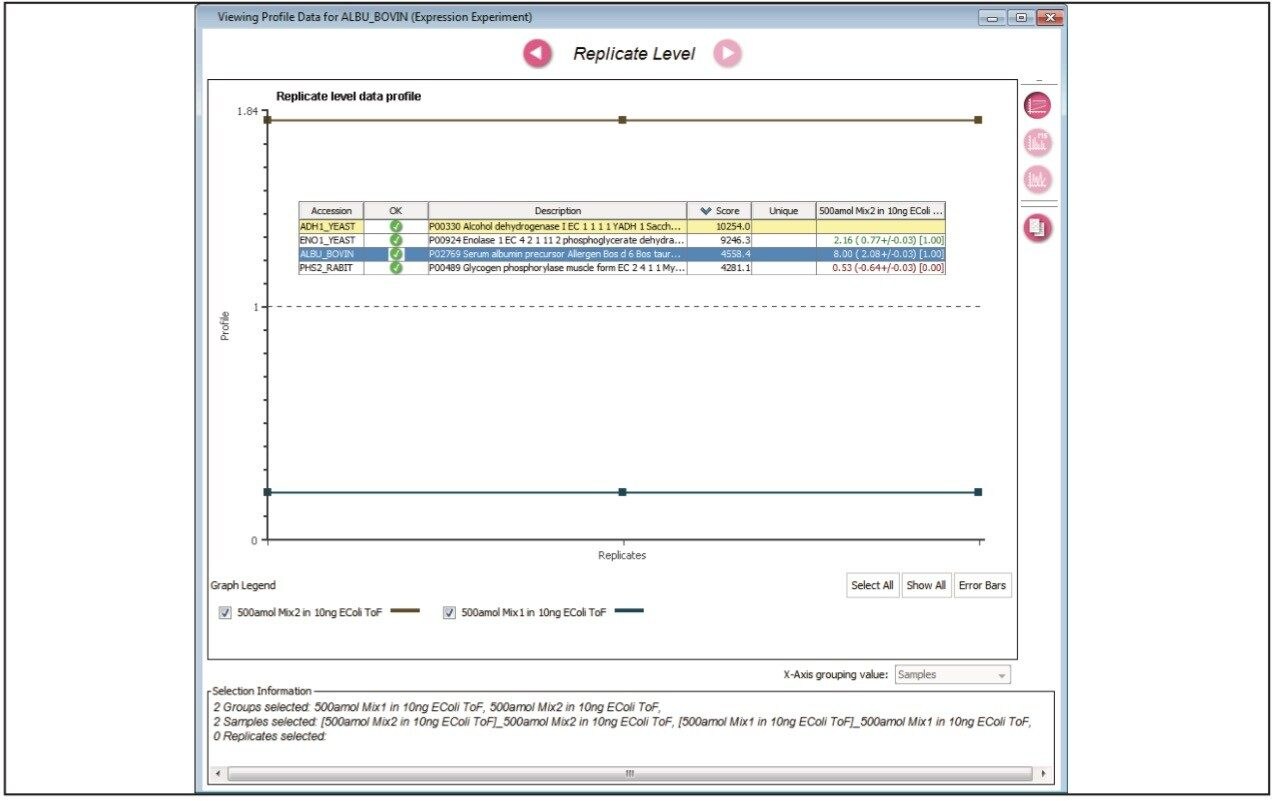
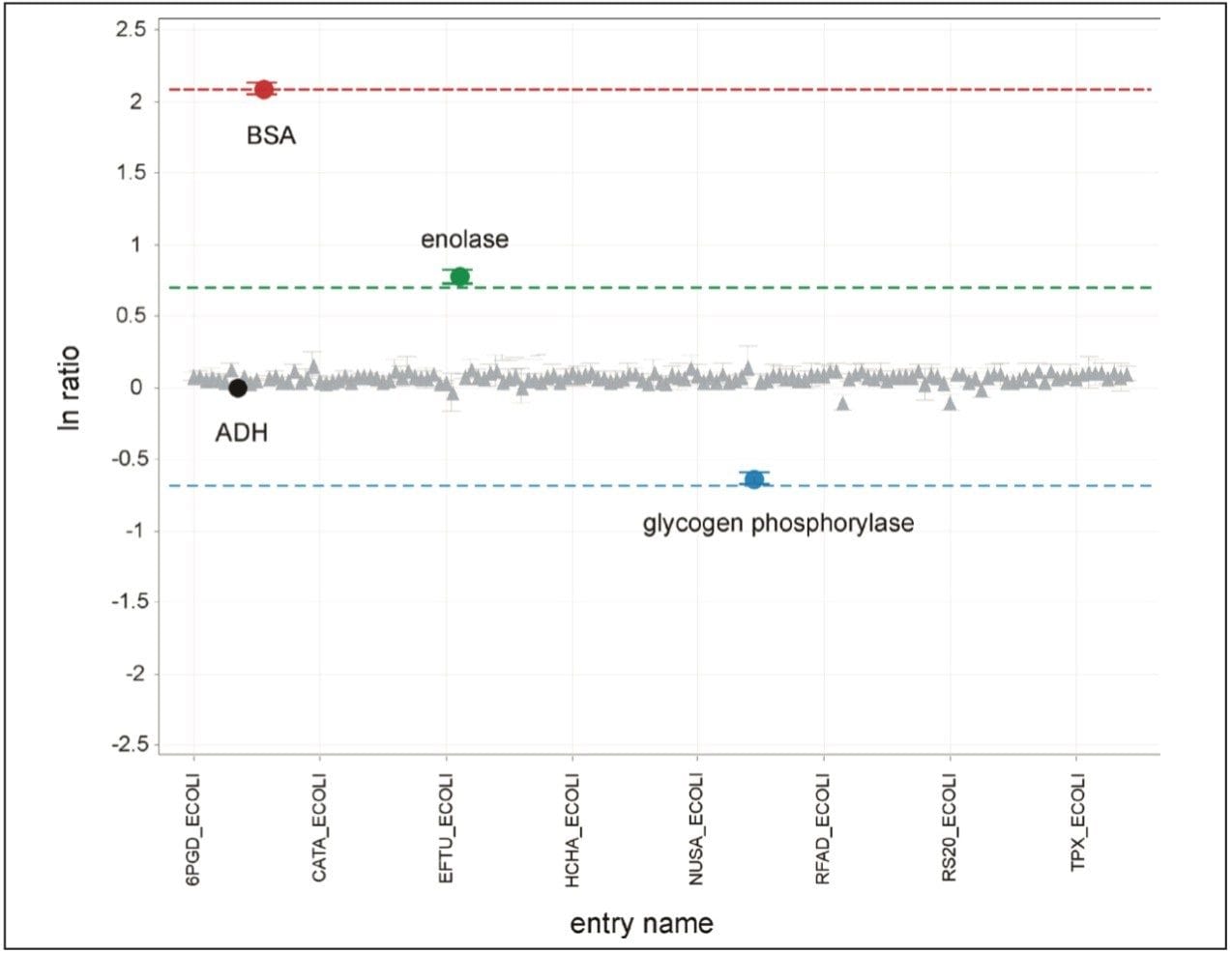
Figure 1 illustrates the qualitative results overview for an LC-MSE acquisition of one of the analyses of the differentially spiked samples. In this particular instance, the on-column amount of highlighted BSA was 4 fmol and the amount of E.coli digest was 10 ng. The results shown in Figure 2 demonstrate the corresponding relative quantification result. A graphical representation for all of the proteins is shown in Figure 3.