Mobility Assisted Data Independent, Relative Label-Free Protein Quantification
This is an Application Brief and does not contain a detailed Experimental section.
Abstract
The quantitative analysis of complex protein mixtures requires tryptic digestion coupled with a protein identification strategy that can accurately identify proteins with high reproducibility. This has often been overlooked and many quantitative measurements have been based upon spurious identifications. The depth to which a complex sample can be interrogated is also crucial as this defines the lowest abundance limit of proteins that maybe quantified. The SYNAPT® G2 HDMS™ System has substantial improvements in performance (mass resolution and accuracy, dynamic range, sensitivity, and mobility resolution). These enable highly accurate protein identification and label-free, relative protein quantification, which we present here.
Benefits
Introduction
Isotope-labeled and label-free protein quantification in increasingly complex proteomics samples is conducted to overcome the bias and difficulty of sample fractionation. This places a demand on the performance of the analytical LC/MS system, requiring the highest obtainable coverage of the proteins present within the sample in order to identify quantifiable, proteotypic information, without increasing the protein false discovery rate (FDR) and false quantification rate (FQR). As such, technology improvements are indispensable. The use of the SYNAPT G2 HDMS System and the results for the analysis of differentially spiked protein standards in a complex cell lysate from Escherichia coli (E. coli) are described.
Results and Discussion
Three replicates of each E. coli sample, differentially spiked with bovine serum albumin (BSA), alcohol dehydrogenase (ADH), Enolase and Glycogen phosphorylase B were analyzed. The nominal ratios were 1:8, 1:1, 1:2, and 2:1, respectively. The peptides were separated and analyzed using a nanoACQUITY UPLC® System coupled with a SYNAPT G2 HDMS. The data were acquired in LC/HDMSE mode – an unbiased mobility assisted TOf acquisition method – an unbiased mobility assisted TOf acquisition method – switching between low and elevated energy on alternate scans and correlating precursor and product ions by means of retention and drift time. Searches and quantification were conducted with ProteinLynx Global SERVER™ v2.5 using a species specific database to which sequence information of the spiked protein was appended.
Figure 1 illustrates the qualitative results overview for an HDMSE acquisition of one of the analyzed samples. In this particular instance, the on-column amount of highlighted Enolase was 12.5 fmol and the amount of E. coli digest equal to 200 ng. The results shown in Figure 2 demonstrate the relative quantification result for BSA. A graphical representation for all quantified proteins, including the protein spikes, is shown in Figure 3.
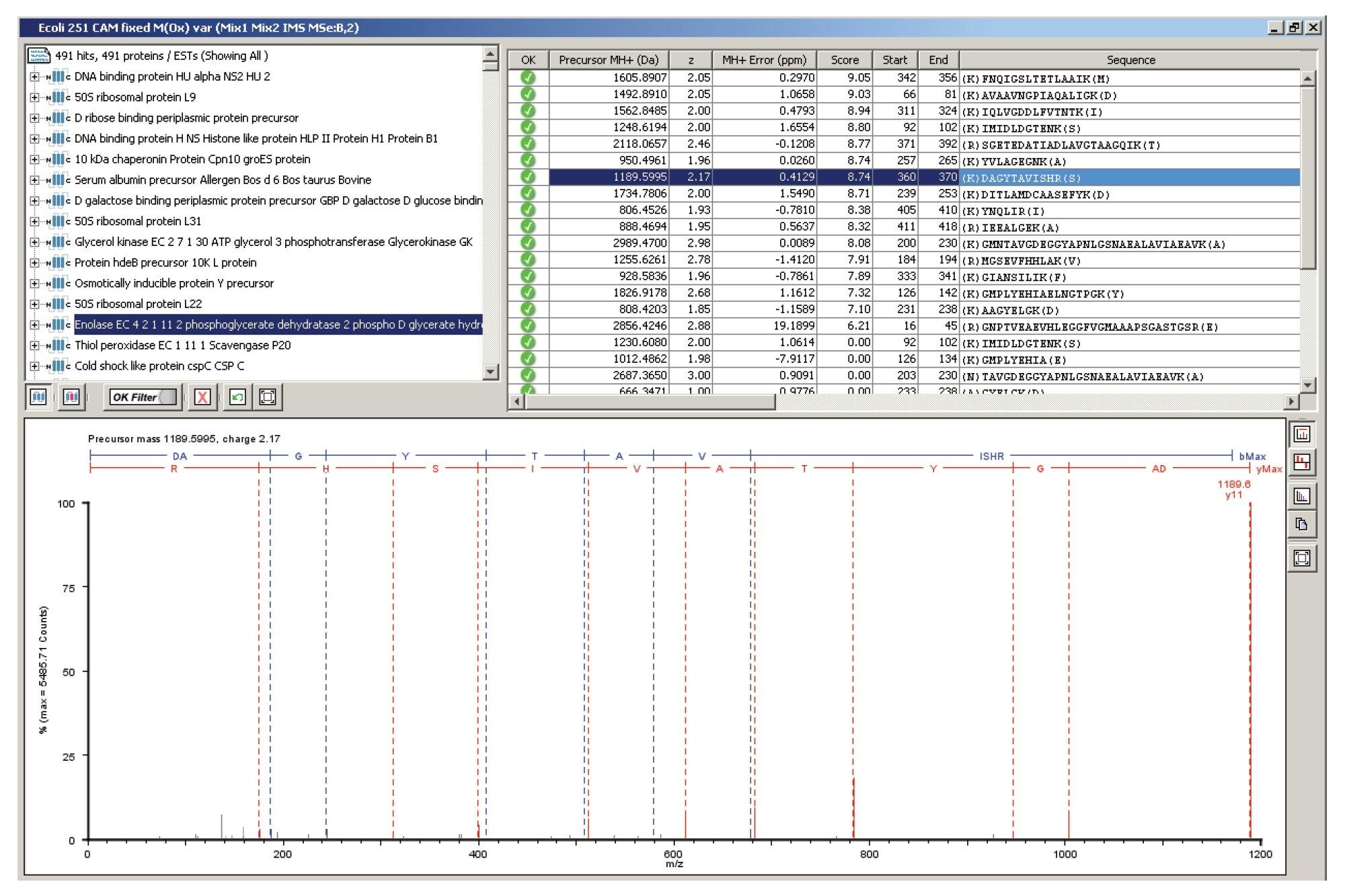
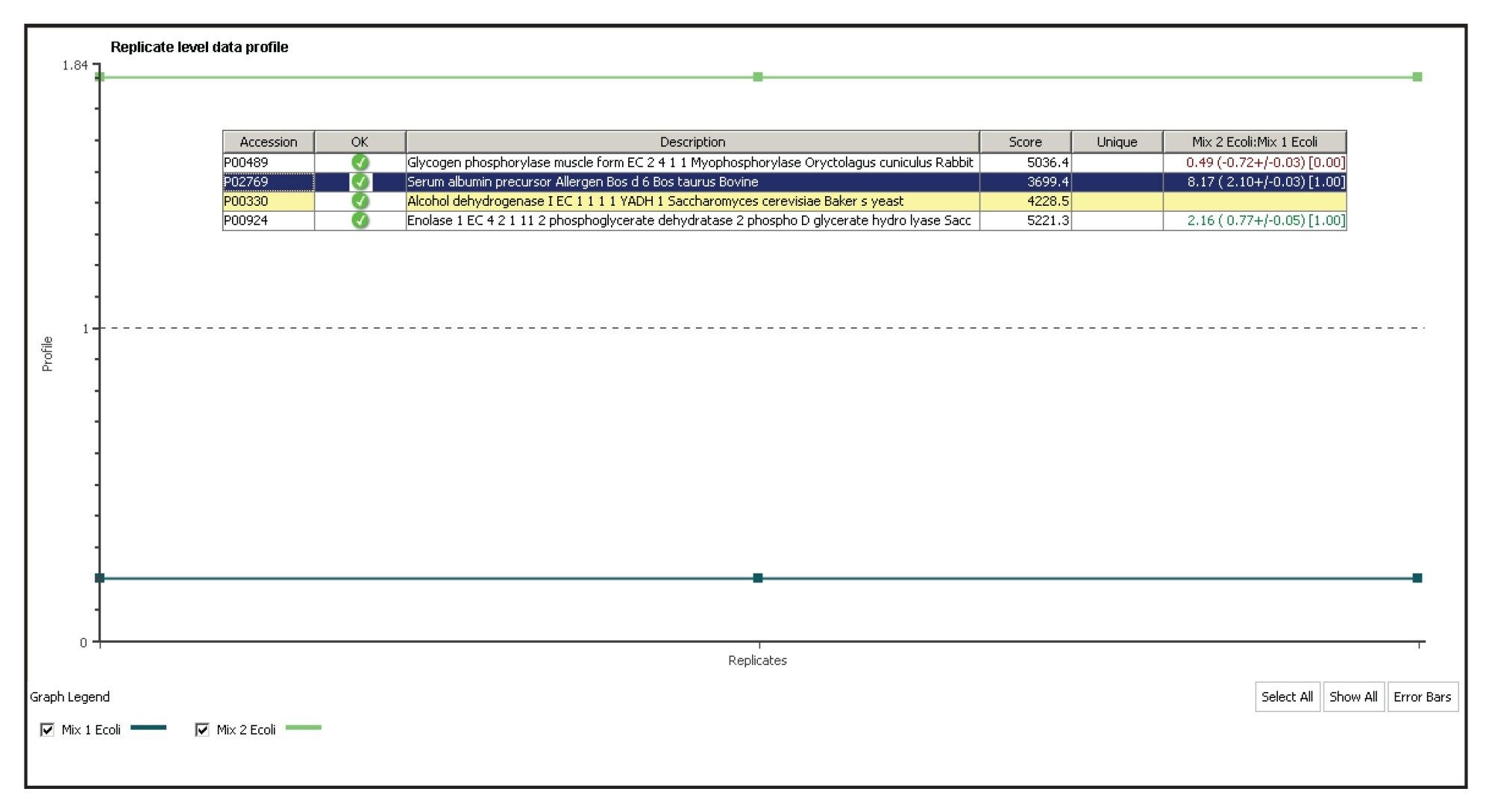
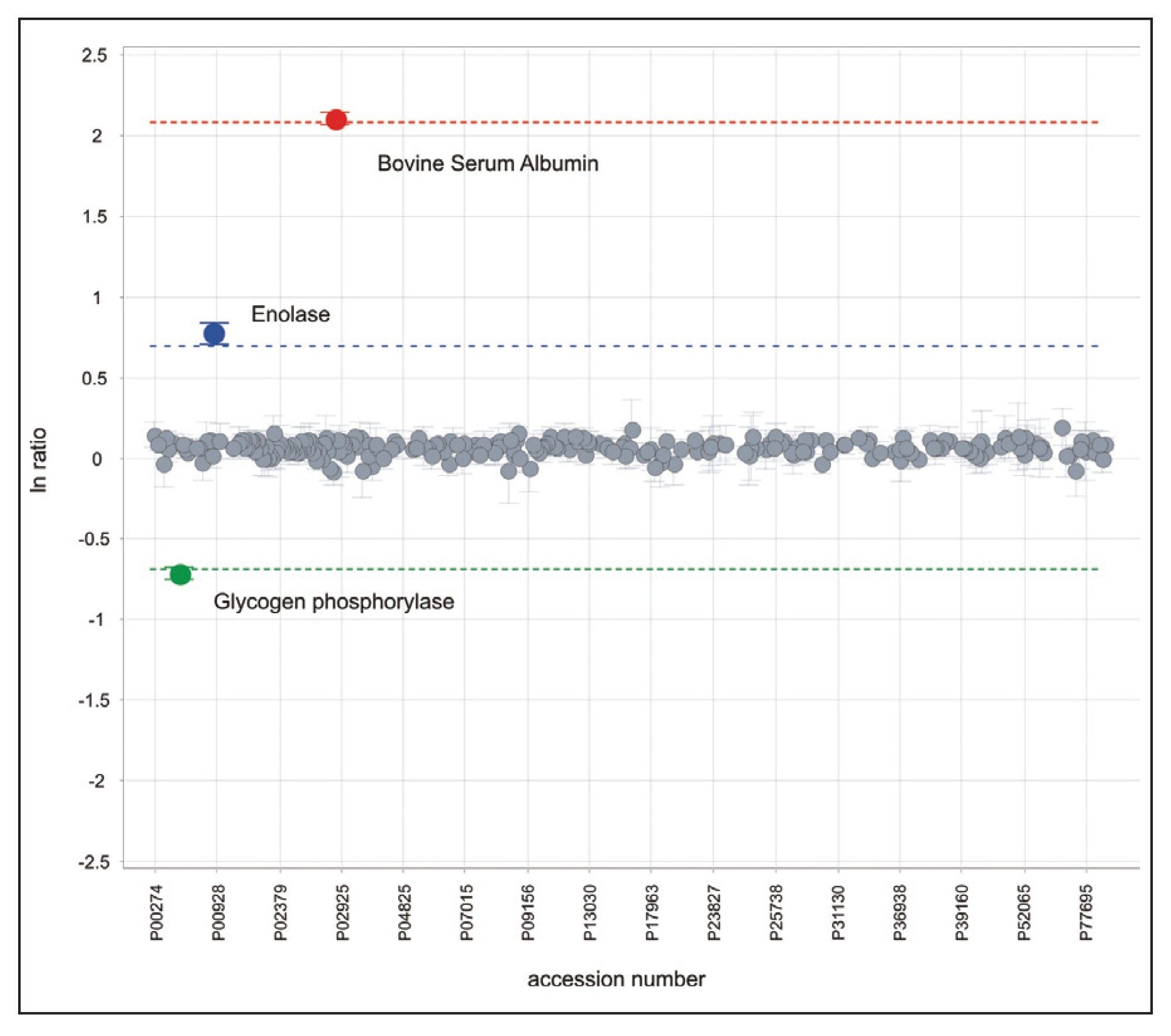
Conclusion
In this technical brief, we demonstrate an ion mobility-enabled data independent acquisition method for quantification of four protein standards, which were spiked into a biological background. The additional specificity of the IMS-enabled experiment provides highly accurate information for qualitative and quantitative experiments. All of the reported protein ratios are within a few percent of their nominal spike values and included in the reported 95% confidence intervals.
Featured Products
720003881, March 2011