Influenza is a viral infection of the respiratory tract. It is one of the leading causes of death in the U.S., killing more than 50,000 people per year.1 Influenza vaccination is a primary prophylactic method and the principal strategy for reducing morbidity and mortality due to seasonal influenza. Vaccines provide protection by neutralizing antibodies to viral hemagglutinin (HA), a protein that plays a critical role in influenza infection.
Licensed, inactivated vaccines for seasonal influenza usually contain a preset amount of HAs – a mixture of H1, H3, and B, the corresponding HA proteins of the three most common viruses – influenza A subtypes H1N1 and H3N2, and influenza B. These HA proteins are glycoproteins, with multiple N-linked glycosylation motifs and multiple glycoforms for each glycosylation site. Detailed characterization and monitoring of glycosylation in HAs is important for both vaccine development and production because of their role in determining the function of influenza binding onto host cells and therefore infection.
Currently, the methods for characterization of N-linked glycosylations include released free glycan analysis2-4 and intact mass analysis.5-7 These methods are useful for analyzing glycoproteins with defined glycosylation sites, such as monoclonal antibodies. Glycan profiling can be performed at the intact protein level (intact mass analysis) or as carbohydrates (free glycan profiling). The glycosylation site can usually be determined by peptide mapping8 after the glycan moieties are enzymatically removed (mostly by PNGase F) because the mass of asparagine (N) residues with glycosylation increase by 1 Da upon deglycosylation.
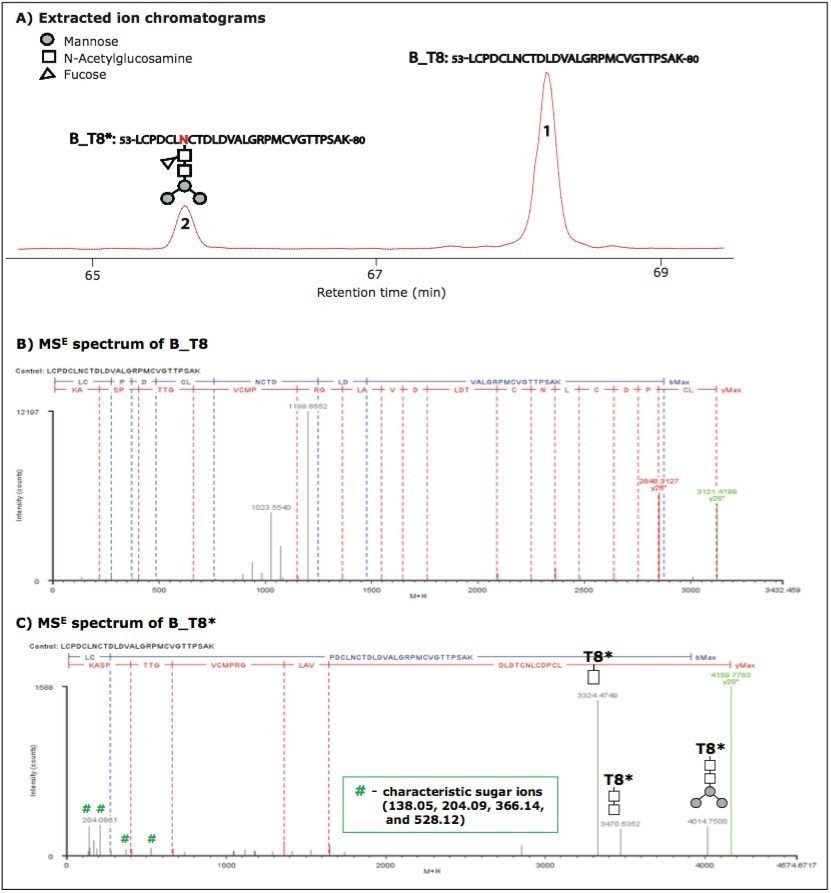
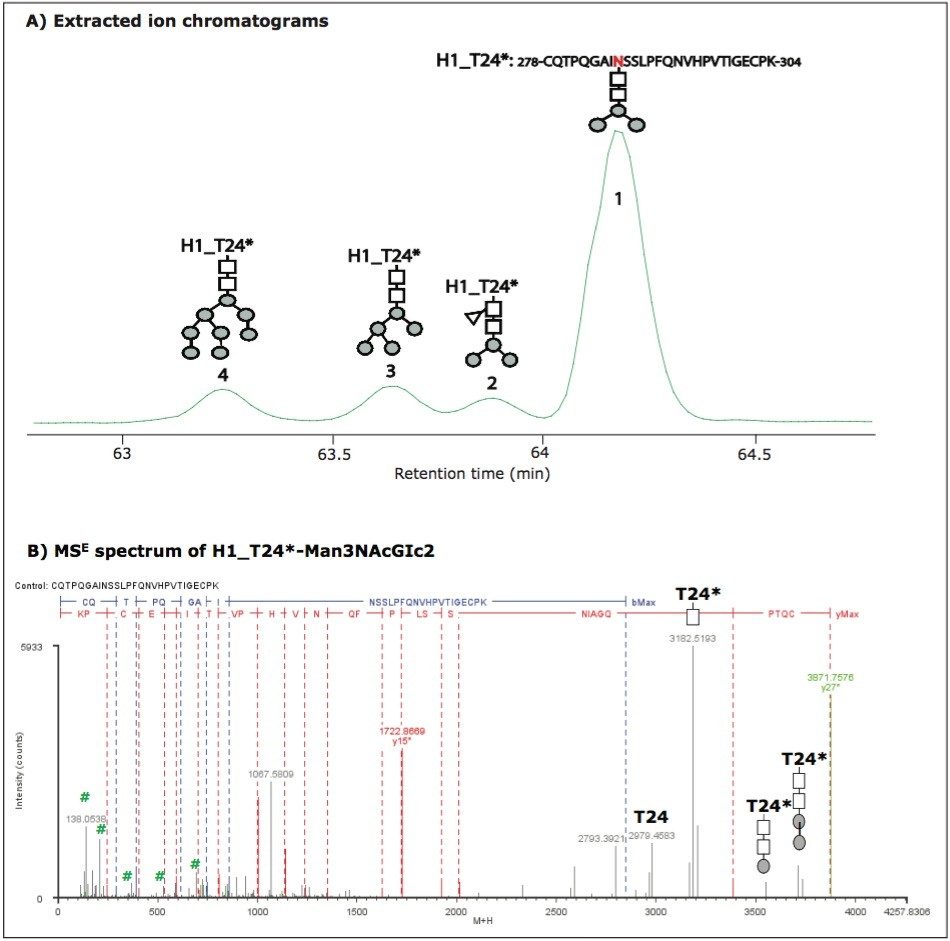
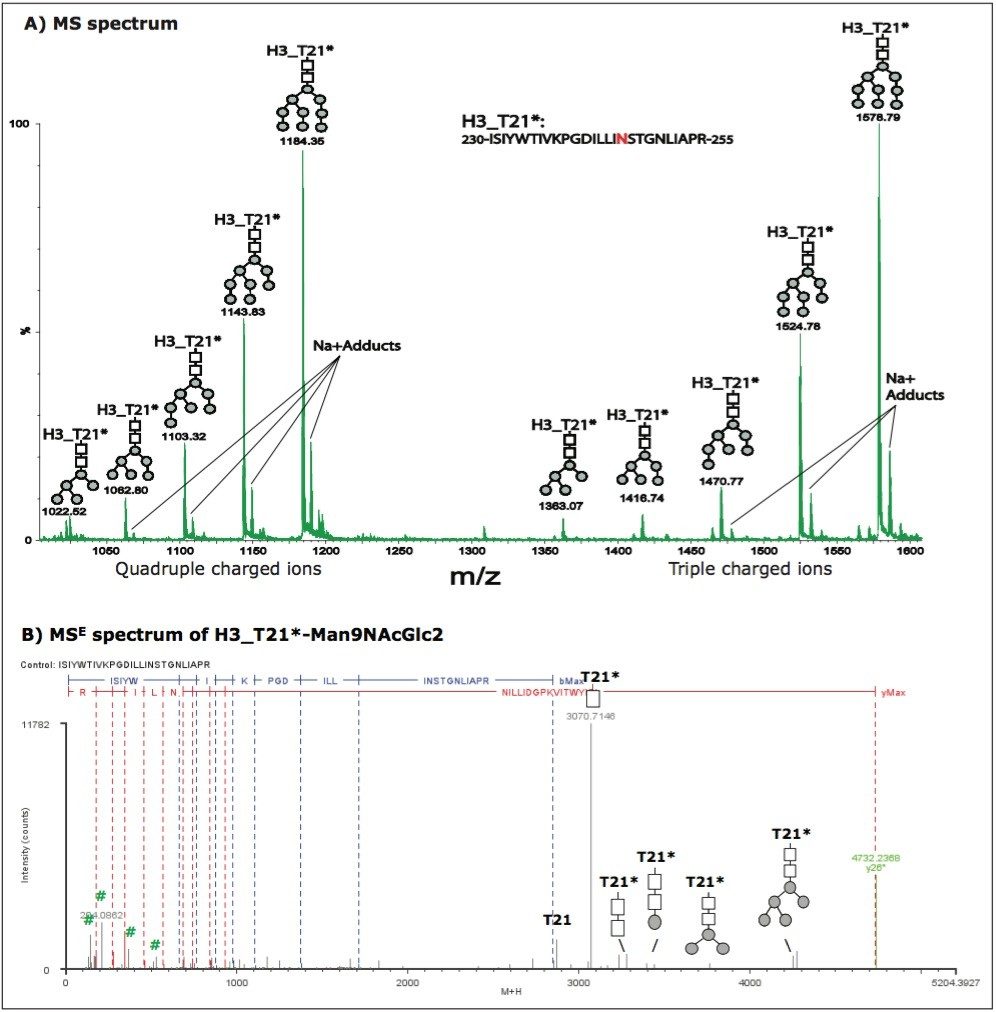
However, it is a challenge for these methods to characterize glycoproteins with multiple glycosylation sites, such as HAs, because it is difficult to distinguish between glycan moieties that link to different sites in a protein. Furthermore, complex samples like vaccines have multiple N sites with an -NXS/T- motif. The determination of N-linked glycosylation sites by peptide mapping may be extremely challenging because modifications of N sites with a 1 Da mass increase could be due to either glycosylation or deamidation.
Using improved resolution of UPLC, four major N-linked glycoforms of a tryptic peptide prepared from a monoclonal mouse IgG1 antibody tryptic digestion were analyzed by LC/UV-MS.9 The experiment demonstrated that glycosylations could be detected and quantified. Both glycosylated site and glycan moiety could be characterized simultaneously.
In previous studies,10-11 we demonstrated that tryptic peptide mapping with UPLC-MSE was capable of separating and characterizing site-specific modifications such as N-deamidation and M-oxidation in an unbiased manner.
In this application note, we demonstrate that UPLC-MSE is capable of separating and characterizing N-linked glycosylations of HA proteins in a recombinant influenza vaccine candidate expressed from insect cell-baculovirus expression vector system (BEVS). Glycopeptides and glycoforms are separated by an ACQUITY UPLC System at the peptide level, and are detected online by a SYNAPT MS System. The UPLC-MSE data are processed by BiopharmaLynx Software to report N-linked glycosylation information. The method offers a way to improve the characterization as well as reduce the amount of time spent on data processing. Furthermore, having a general technique that can be applied to such problems opens this type of analysis to non-experts and could benefit an organization by streamlining their work.