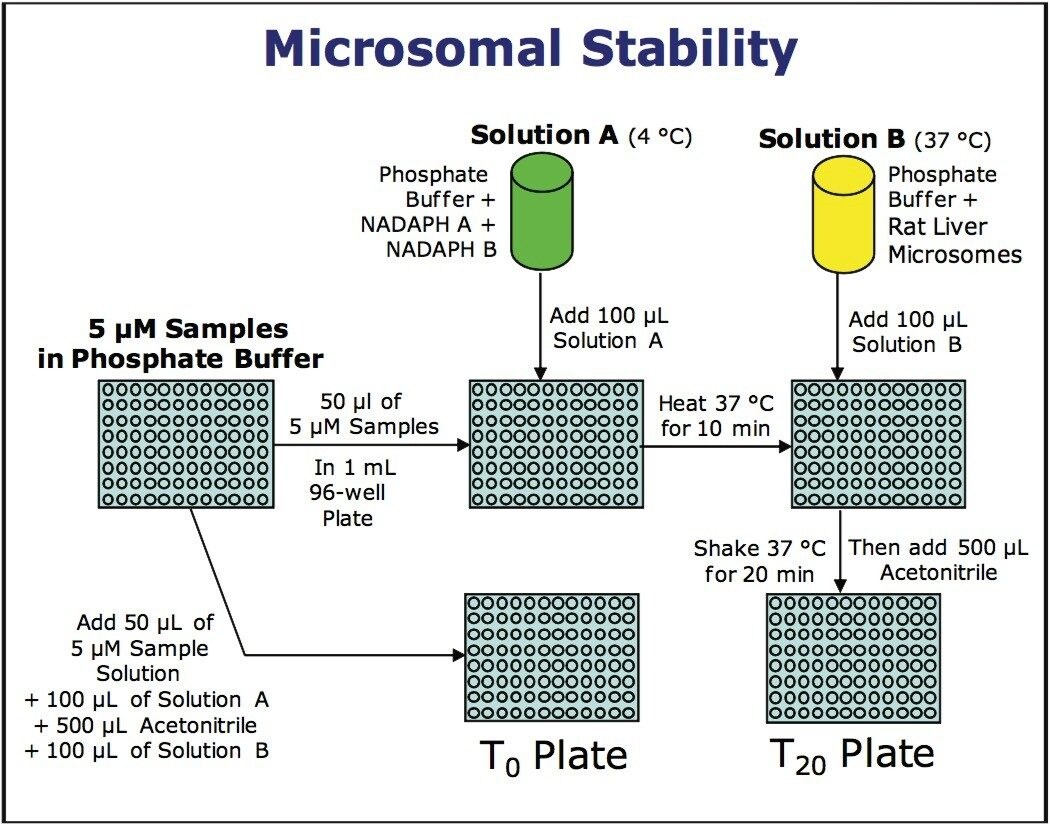
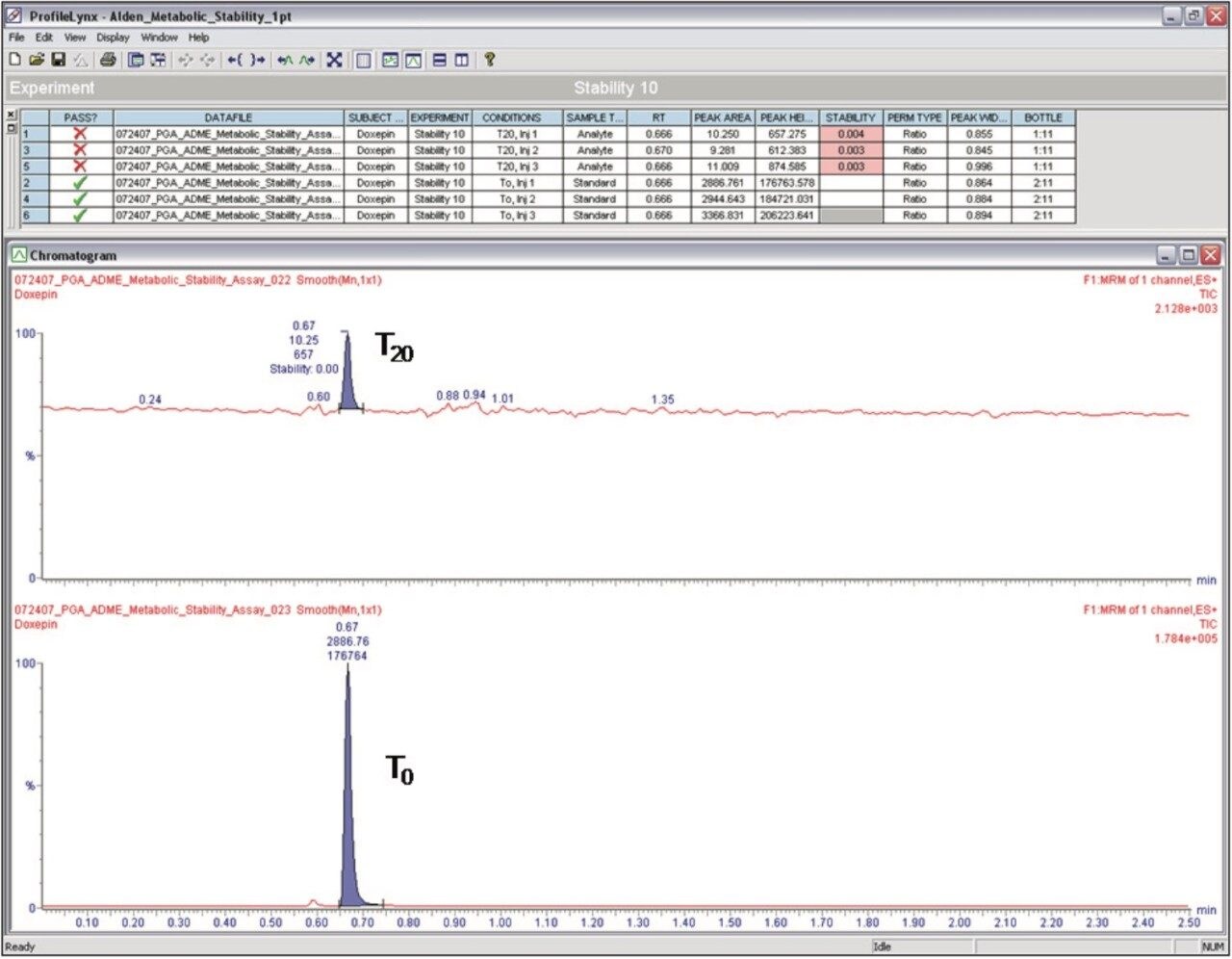
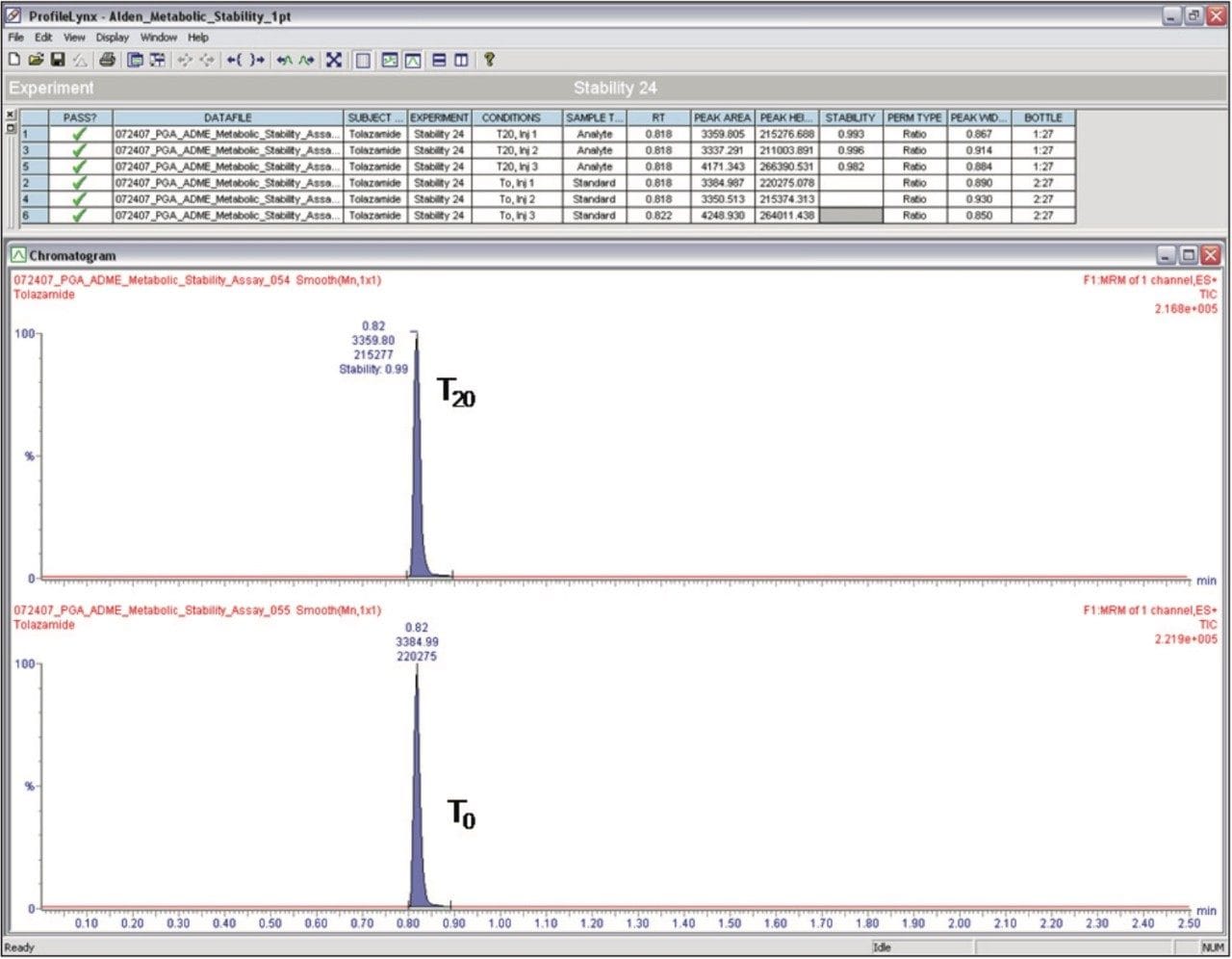
Preparation of Solutions
Solution 1 - 0.1M Potassium Phosphate Monobasic
13.6 g of potassium phosphate monobasic (KH2PO4) was weighed and transferred to a 1-L volumetric flask, diluted to volume with Milli-Q water, and mixed thoroughly.
Solution 2 - 0.1M Potassium Phosphate Dibasic
17.42 g of potassium phosphate dibasic (K2HPO4) was weighed and transferred to a 1-L volumetric flask, diluted to volume with Milli-Q water, and mixed thoroughly.
100 mM Phosphate Buffer
190 mL of solution 1 was combined with 810 mL of solution 2, mixed thoroughly and filtered through a 0.2-μm nylon filter. The pH was adjusted to 7.4 with phosphoric acid.
Solution A - Cofactor Solution
10.938 mL of 100 mM phosphate buffer was added to each of two ~45 mL plastic culture tubes (VWR), and the tubes were put in an ice bath at 4 °C. 1.609 mL of cofactor A and 322 μL of cofactor B were added to each tube, which were then mixed and kept at 4 °C.
Solution B - Rat Liver Microsome Solution
10.938 mL of 100 mM phosphate buffer was added to each of two ~45 mL plastic culture tubes (VWR), and the tubes were warmed to 37 °C on a Boekel Scientific Jitterbug shaker. 729 μL of rat liver microsomes were added to each of the tubes and mixed thoroughly.
Sample Dilution Buffer
10 mL of 0.5 M EDTA solution was added to 990 mL of the 100 mM phosphate buffer and was mixed thoroughly.
5 μM Sample/Standard Plate
Standards and samples were prepared at a concentration of 0.5 mM in DMSO. 10 μL of each of these samples (and standards) were transferred to a 2-mL, 96-well plate. To each well was added 990 μL of sample dilution buffer and the solutions were mixed thoroughly.