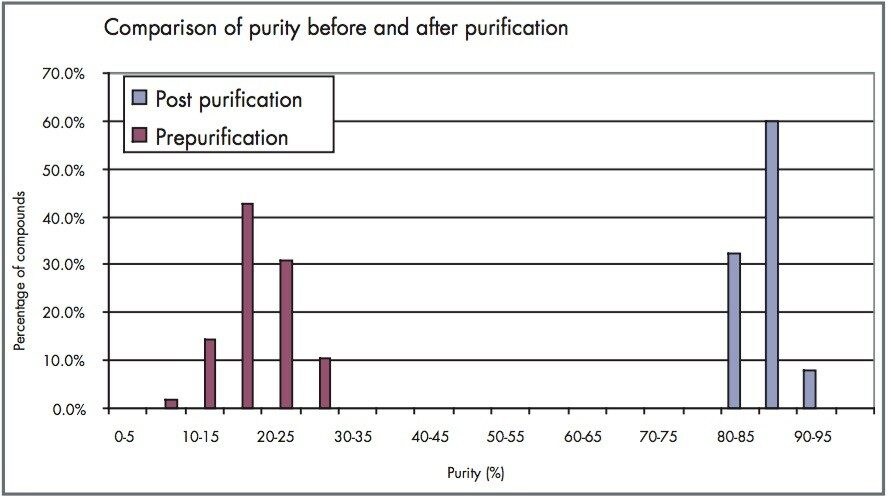
The primary objective of this experiment was to increase the throughput of the purification process, while maintaining acceptable sample recovery and purity. To this end, chromatographic conditions were developed to allow for the fastest run time and still meet 85% purity criteria.2,3 In addition, minimal user intervention in the process was required to maximize efficiency.
Using the Waters Purification Factory, it was possible to purify 3940 samples in 10 days. Inject-to-inject cycle times were 11.5 minutes long and the system only ran 18.5 hours a day. If the system were to be run at full capacity, over 500 samples could be purified every day with this gradient length. Intervention was minimized due to the robustness of system operation and ruggedness of the preparative columns.
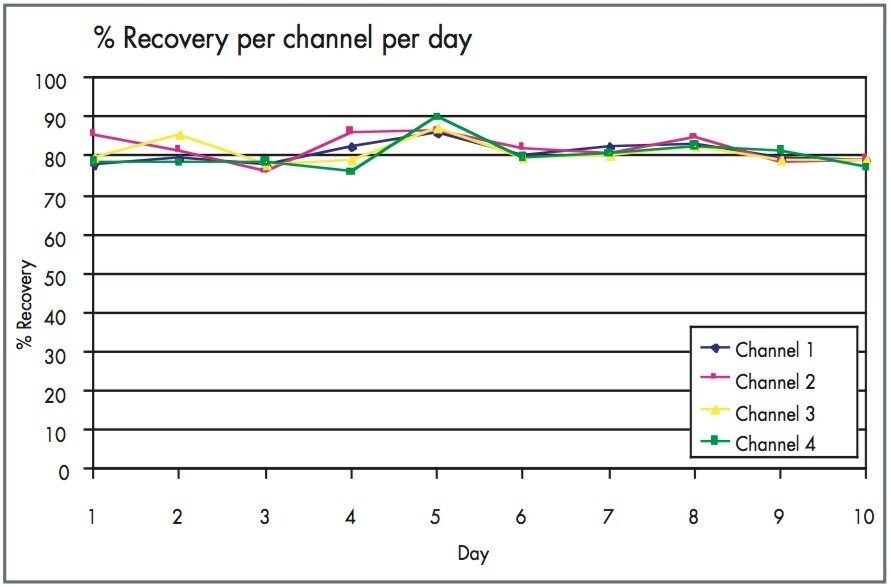
One of the major problem areas of high throughput preparative chromatography is the potential for blockage of either the column or the splitter due to the high concentration of material being injected. Using four 2525 pumps alleviates this issue. If one channel does have a problem, the fractions from the other channels are not affected. In addition, because any pressure changes are not spread over the other channels, blockages are detected earlier by the system and the system can safely shut down.
Four pumps also have another throughput enhancement. They allow for the possibility to set up different gradients on each channel, increasing the number of different compounds and associated chromatography that can be analyzed. It allows for the running of shallower gradients that focus in on the peak of interest, decreasing gradient run time. By using the AutoPurify feature of FractionLynx, this capability can even be automated.4
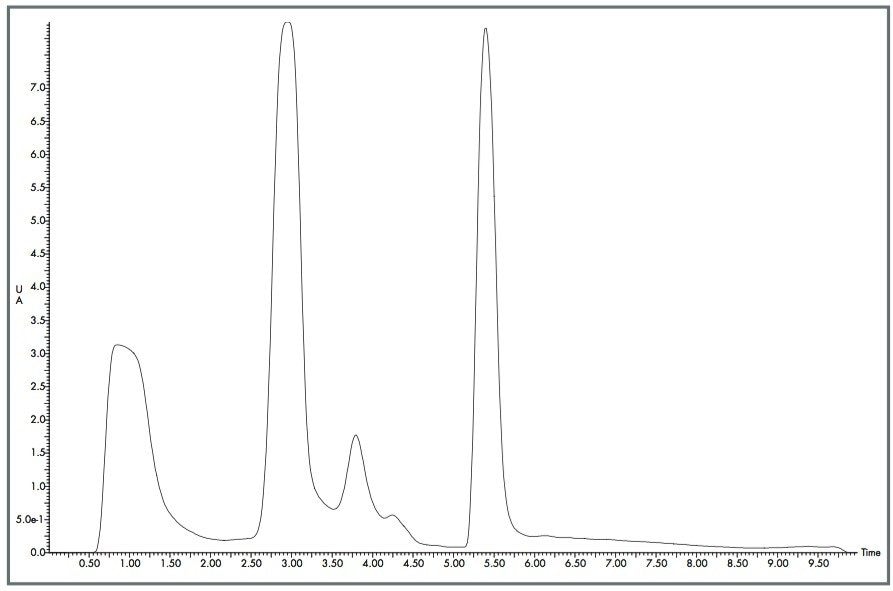
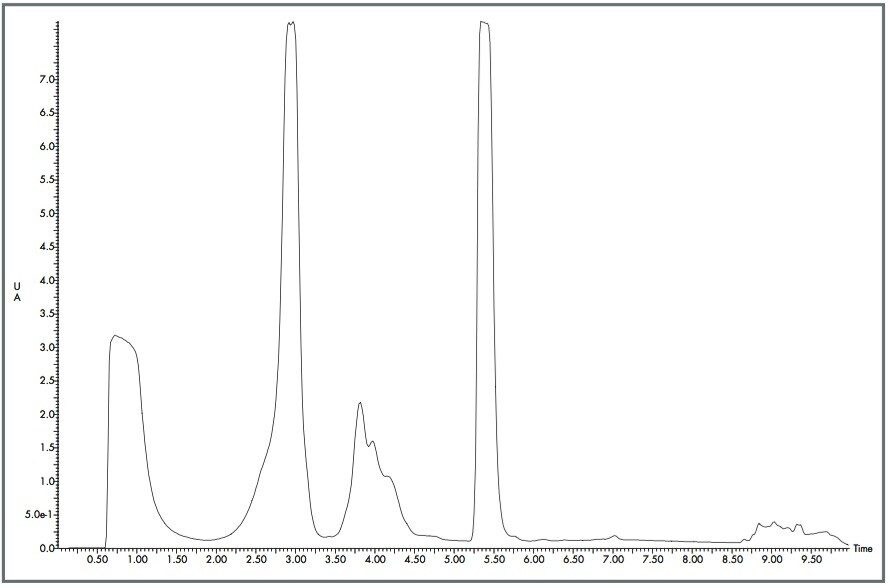
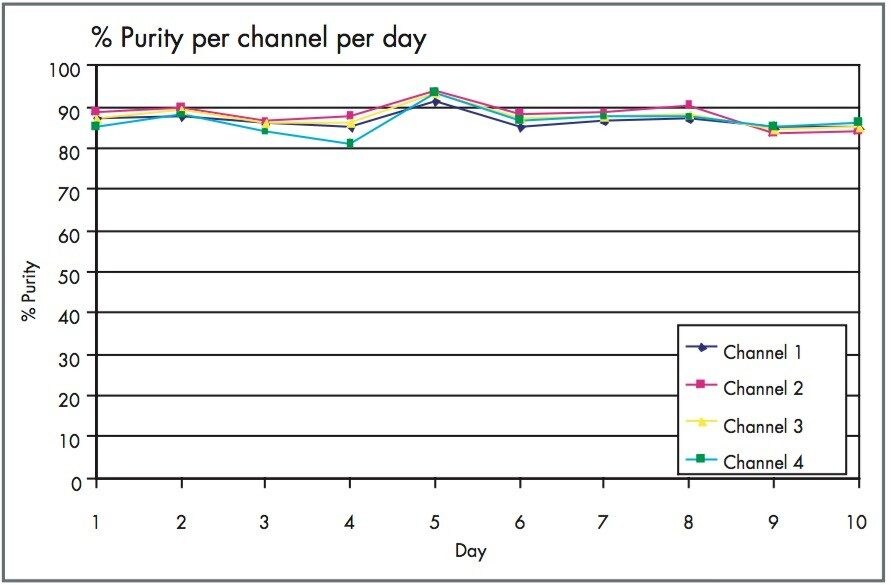
The Optimized Bed Density design of the Waters XTerra Prep Columns allowed 30 mg of material to be loaded onto each column 960 times with little change in peak shape, as shown in Figures 1 and 2. Even with nearly 1000 injections per column, the columns showed acceptable performance and could be used for additional studies.5