Rapid, Efficient, and Simple Sequence Confirmation of Synthetic Oligonucleotides Using Confirm Sequence With Targeted and Data-Independent Fragmentation Data
Abstract
This application note demonstrates the efficiencies gained using the waters_connect™ CONFIRM Sequence application software for sequence confirmation of synthetic oligonucleotides when compared to a manual approach for both QTOF (Vion™ Mass Spectrometer) MS/MS and benchtop TOF (BioAccord™ Mass Spectrometer) MSE (DIA) data.
Benefits
Introduction
Synthetic oligonucleotides have emerged in recent years as a powerful alternative to small molecule and protein therapeutics.1 Increasing amounts of oligonucleotide drugs reaching the market require analytical methods that can provide reliable and robust quality control.2,3 The two major categories of synthetic oligonucleotide reaching the market are antisense oligonucleotides (ASOs) and silencing RNAs (siRNAs).4,5 Both modalities typically employ oligonucleotides smaller than 50 monomers in length.
Mass Spectrometry has proven to be the gold standard approach for characterizing such molecules, especially when modified nucleosides and backbones are utilized.6-8 Characterizing the sequence of a synthetic oligonucleotide via targeted MS/MS fragmentation is the preferred approach with well understood and annotated fragmentation pathways.9 Recent investigations into the relationship between oligonucleotide size, precursor charge state and collision energy allow for optimized conditions to generate the sequence confirmation required for regulatory submission.10
Two key limitations to this workflow are the manual processing of resulting complex datasets, as well as the limited availability of experienced analysts required to generate high quality MS/MS fragmentation on the mass spectrometers typically used for discovery and early development. Both of these limitations restrict the downstream adoption of MS/MS based sequence confirmation in manufacturing and quality control laboratories.
Here it is demonstrated that the rapid turnaround of high-quality sequence confirmation data on CpG7909, a modified synthetic oligonucleotide therapeutic, using the waters_connect™ CONFIRM Sequence application software.11 Further demonstration of the potential for routine quality control of synthetic oligonucleotide manufacturing with a benchtop BioAccord™ System (UPLC™-TOF-MS) that was designed for users with limited MS expertise. These results are compared with MS/MS data taken from a QTOF MS (Vion™ MS, an instrument typically used for oligonucleotide characterization.
Manually predicting the expected fragment ions of a synthetic oligonucleotide is challenging. The library features within the CONFIRM Sequence application allow users to create, manage and store custom oligonucleotide sequences using structural information down to the subcomponent level (bases, sugars and linkers). The default monomer entries that come preinstalled with the application are sufficient to create CpG7909, but the library is highly customisable in monomer creation and setting custom fragmentation rules if required.
Several hours of labor-intensive manual processing was reduced to minutes of automated data processing. Reviewing the results was made simple with graphical representation, automated flagging of problematic data areas, and efficient curation of results. The use of a bespoke Targeted Isotope Clustering algorithm in CONFIRM Sequence provides high confidence with detailed fragment ion matching annotated directly on the raw data.
Experimental
Sample Description
CpG7909, sequence dT*dC*dG* dT*dC*dG* dT*dT*dT* dT*dG*dT* dC*dG*dT* dT*dT*dT* dG*dT*dC* dG*dT*dT (d refers to a deoxyribonucleic acid, * refers to a phosphorothioate backbone modification) was synthesized in house at BioSprings via solid phase synthesis. CpG7909 was diluted with water to a final concentration of 0.5 mg/mL.
LC Conditions
|
LC system: |
ACQUITY™ UPLC™ binary solvent manager |
|
Optical detection: |
TUV |
|
Column(s): |
ACQUITY UPLC Oligonucleotide BEH™, 130 Å, C18 1.7 µm 2.1 x 100 mm |
|
Column temperature: |
60 °C |
|
Injection volume: |
2 µL |
|
Flow rate: |
0.3 mL/min |
|
Mobile phase A: |
7 mM TEA and 80 mM HFIP dissolved in water |
|
Mobile phase B: |
3.5 mM TEA and 40 mM HFIP in 50% methanol 50% water |
|
Gradient: |
The LC gradient used was as follows: initial conditions 10 %B buffer for 0.2 min, from 0.2 min to 2.0 min from 10 %B to 100 %B, from 2.00 to 2.20 min 100 %B, from 2.20 to 2.30 min from 100 %B to 10 %B and from 2.30 min to 3.00 min 10 %B. Column temperature was 60 °C with a flow of 0.3 mL/min. |
MS Conditions
The BioAccord™ System RDa™ was used in full scan with fragmentation mode in the low mass range acquisition range from 50–2000 m/z. ESI Polarity was set to negative with a scan rate of 1 Hz. Cone voltage was adjusted to 50 V and the fragmentation cone voltage was varied, starting from 60 or 70 V ramped up to 100 to 120 V. The capillary voltage was set to 0.8 kV, the desolvation temperature to 550 °C and the desolvation gas flow to 800 L/h.
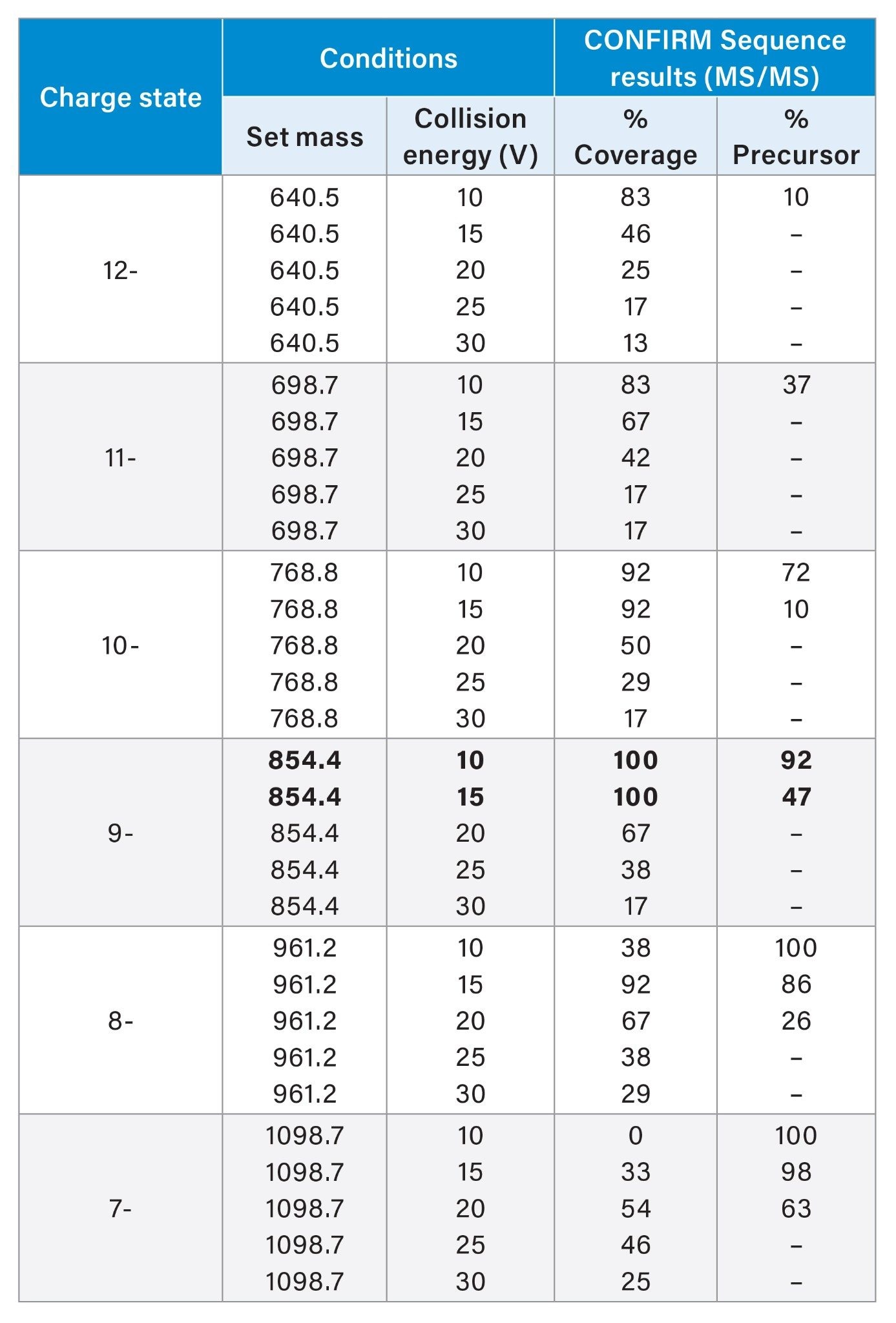
The Vion IMS QTOF was used in ESI negative polarity with standard transmission in the sensitivity mode. Source type was set to ESI with a source temperature of 120 °C, a desolvation temperature of 400 °C, a desolvation gas flow of 800 L/h, a cone gas flow of 50 L/h, a capillary voltage of 2.0 kV, a sample cone voltage of 60 V and a source offset voltage of 100 V. MS/MS acquisition was set in the range of 50 to 2000 m/z, a scan time of 0.5 s and low precursor resolution. The following precursor ions (values in m/z) were selected: 640.5 (charge state: 12-), 698.7 (charge state: 11-), 768.8 (charge state: 10-), 854.4 (charge state: 9-), 961.2 (charge state: 8-) and 1098.7 (charge state: 7-). The collision energy was varied from between 10, 15, 20, 25, and 30 V for each precursor ion.
Processing Settings
|
CONFIRM Sequence method settings (MS/MS) |
|
|
PPM error: |
20 |
|
Intensity cutoff (counts): |
20 |
|
Isotope similarity threshold: |
65 |
|
Isotope intensity threshold: |
65 |
|
CONFIRM Sequence method settings (MSe) |
|
|
PPM error: |
30 |
|
Intensity cutoff (counts): |
200 |
|
Isotope similarity threshold: |
65 |
|
Isotope intensity threshold: |
55 |
Results and Discussion
Sequencing of oligonucleotides using QTOF MS involves charge state selection and collision energy screening for optimal results. Depending on the charge state, the collision energy needs to be optimized to avoid low energy partial fragmentation and high energy over-fragmentation (internal breaks of the sequence and fragmentation of fragments).
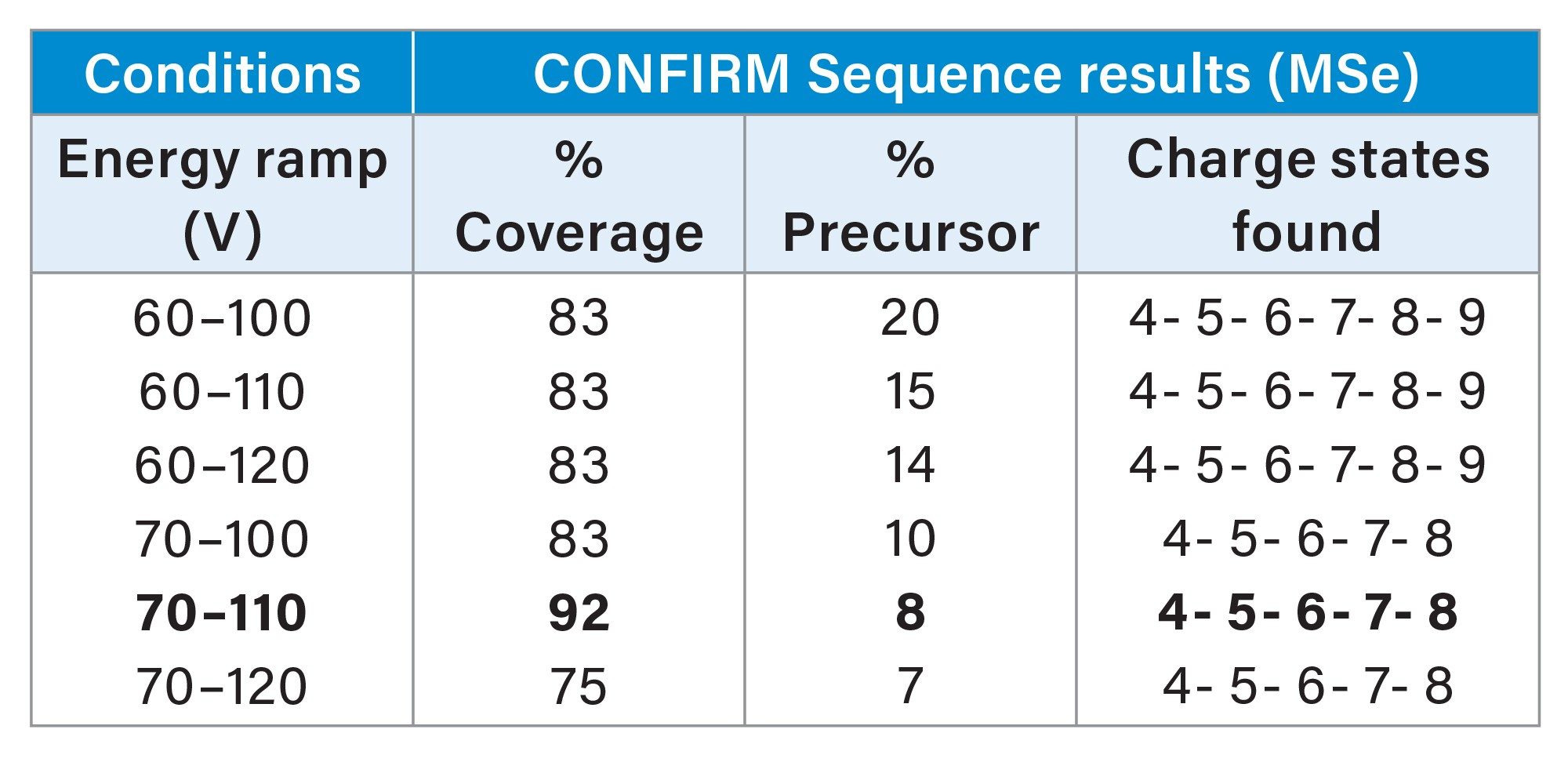
For our test molecule CpG7909, an immunostimulatory adjuvant synthetic oligonucleotide therapeutic, the observations of increased energies required for lower charge states made by Gawlig and Rühl (2023) are also seen here for both MS/MS (Table 1) and MSe (Table 2) analysis.10
An additional strategy for fragmentation tuning is to target the amount of residual precursor that is present in the fragmentation data. Without residual precursor signal present, it is likely that the molecule has been over fragmented, or that the charge state is unstable in the given conditions. A reduction of %precursor value displayed in CONFIRM Sequence correlates with increasing collision energies in all cases and can also indicate that increased collision energy has caused over fragmentation. Taken together, the presence of a precursor with the highest collision energy for a given charge state can indicate the optimum settings, and the %precursor metric can be used to obtain this optimized result.
With MSe fragmentation, the highest collision energy that provides the minimum amount of residual precursor (5–10%) is optimal in the data shown here. With MS/MS fragmentation the optimum conditions are charge state dependant. Collision energies that show decreased sequence coverage and %precursor compared to a lower energy on the same charge state is an indication of over fragmentation. Optimising the conditions for an MS/MS acquisition should attempt to increase the collision energy until the %precursor is below 60%, and continue to increase the energy until the lowest %precursor is obtained that gives increased sequence coverage compared to a lower energy.
Manual processing vs CONFIRM Sequence
Reviewing data from a manual workflow is time consuming and can be prone to user error. The review steps in CONFIRM Sequence are designed to present sequence coverage in a way that is easy to understand and provides features for manually reviewing each fragment ion match directly annotated on the raw data.
Data processing in CONFIRM Sequence is rapid (less than five minutes to process all the data shown here), and can be pre-configured by an expert user to allow for less experienced users to process large datasets under the expert’s supervision. Furthermore, the waters_connect™ Software platform is compliance-ready, including the necessary reporting, data integrity controls, and method versioning required for regulatory adherence in quality assurance lab operations.
The CONFIRM Sequence app provides high fidelity fragment ion matching and removes isobaric fragment assignments from coverage calculations. This leads to fewer false positive assignments and a reduced burden for manual data verification by a user.
Enhanced matching fidelity
Complex fragmentation data of modified synthetic oligonucleotides across a large mass range can take considerable time to deconvolve (up to 20 minutes per spectrum) and requires some expertise to manually investigate the deconvolved results and assign predicted fragments based on mass.
The CONFIRM Sequence app employs an intelligent Targeted Isotope Clustering algorithm to alleviate the challenges of a manual and untargeted approach. CONFIRM Sequence processes data in a targeted fashion, where the predicted isotope clusters for each fragment ion are searched directly against the raw data and matched based on a variety of scoring metrics. Matches to the raw data can be reviewed in the application to support fragment ion assignments.
Furthermore, a set of logical rules are applied that govern how a set of fragment ions can be used to confirm a monomer assignment. One example of this is with the use of base loss fragmentation ions (e.g a-b-ions), where an ion has undergone gas phase ion/ion rearrangements post collision induced dissociation fragmentation that results in a loss of the base component. Because of this loss, it is not reasonable to use only the base loss ion to confirm the presence of a monomer that is directly 5’ to the fragmentation site. The logic in CONFIRM Sequence dictates that it should be used in the n-1 series of fragment ions instead (e.g. an a-b3 ion should not be used to confirm the presence of the 3rd monomer, instead it should be used in the [x-]2 ion series). This assignment logic used in CONFIRM Sequence is designed to ensure that the monomers confirmed within the application are based on predictable fragmentation modelling and observable data. There are help files in the software that explain each of the monomer confirmation rules in more detail.
Ambiguities
CpG7909 has various predicted fragment ions that are isobaric, and using them to confirm multiple sections of the sequence would be disingenuous. CONFIRM Sequence flags these ambiguities and removes them from the initial sequence confirmation set, but provides users the option to add them back in when reviewing the data. As an example, we added back in the w1- ion, which is isobaric with d1- due to the 5’ end and 3’ end monomers being identical. In this case because these monomers are both at the end of the sequence, we reason that allowing for this ambiguous match is acceptable. However, there are multiple isobaric fragment ions within the CpG7909 sequence that cannot be reasonably used to confirm the sequence, making parts of the sequence difficult to cover. This does however highlight the hazard of performing manual sequence confirmation work, where these ambiguities are less easily identified.
Comparing targeted MS/MS to data-independent acquisition (MSe)
CONFIRM Sequence is able to demonstrate 100% sequence coverage in a single MS/MS fragmentation spectra and 92% using data-independent fragmentation based acquisition (DIA) on the BioAccord System. The benefit of MSe (DIA) acquisition is a simplified setup that fragments all precursor ions with a collision energy ramp designed to match the optimized CE settings which have to be pre-determined using targeted MS/MS acquisition. This removes the need to pre-select target precursor charge states and the experimental design to optimize the collision energy.
To assist the optimization of MS/MS experiments the CONFIRM Sequence App presents results for each MS/MS acquisition together in an overview table (Figure 1), where the combined sequence coverage is displayed alongside a table which displays the individual contributions from each spectra. This allows for instant determination of the optimum fragmentation conditions across all data, and provides insight on which charge state requires more / less energy to achieve optimum conditions.
Reviewing the fragmentation results for individual analyses in the CONFIRM Sequence app is streamlined with annotated spectra for each fragment ion assignment, which can be navigated through and rejected if necessary (Figure 2). Ambiguous fragment ion assignments are also flagged for review.
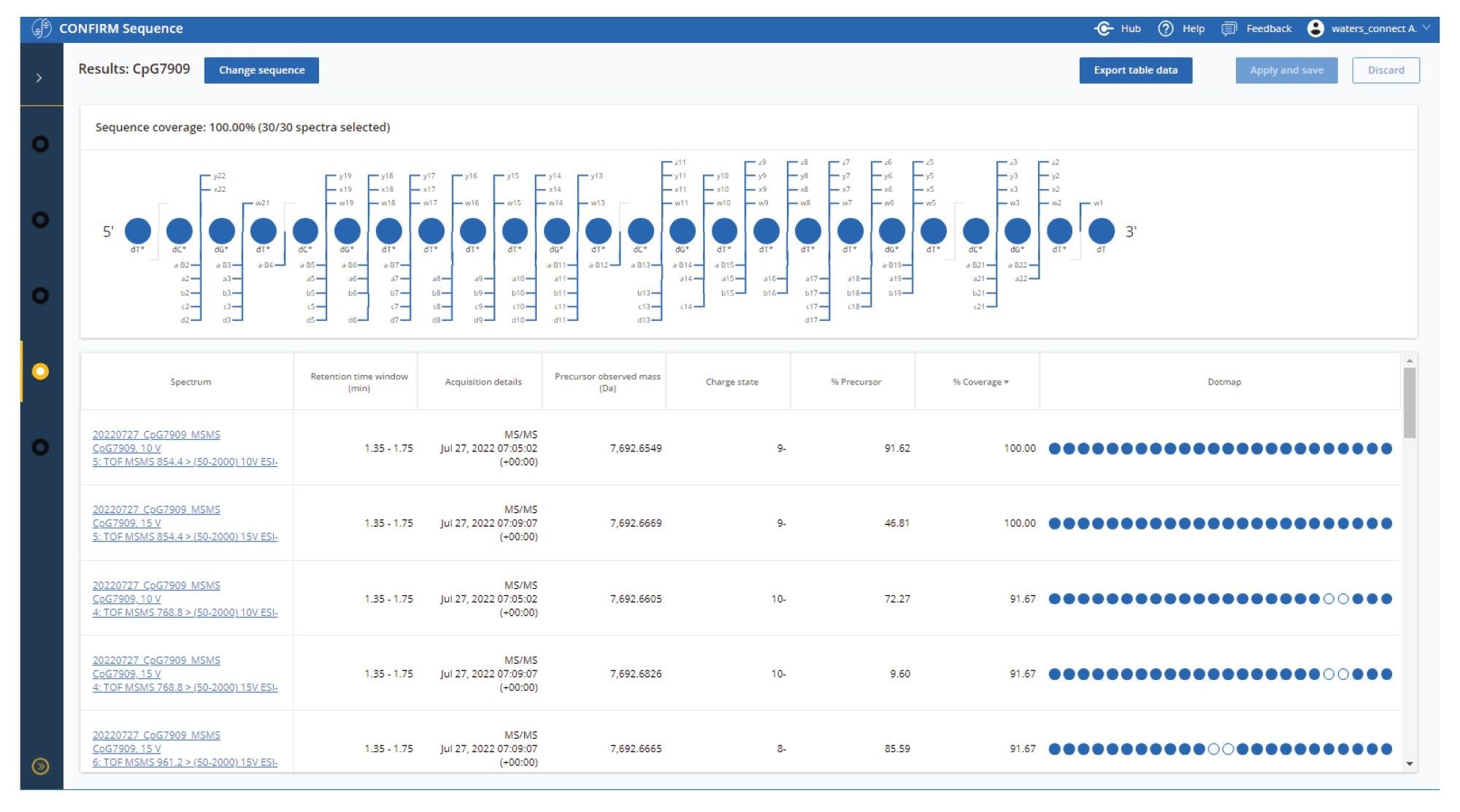
Figure 1. A screenshot of the CONFIRM Sequence application results overview page, showing the results . The data shown here is the results for an MS/MS experiment on CpG7909 with targeted precursors (m/z) at 640.5 (-12), 698.7 (-11), 768.8 (-10), 854.4 ( -9), 961.2 ( -8), and 1098.7 (-7). The collision energy was varied from between 10, 15, 20, 25 and 30 V for each precursor ion. An overview of combined sequence coverage is displayed in a Dotmap™ view, where the sequence is represented with colored orbs and fragment ion ‘legs’, indicating that the monomer is confirmed (blue orb) based on the fragment ions that have been matched in the raw data. Unconfirmed monomers are represented with a white orb.
The table presents results for individual MS/MS spectra and includes information on the Injection / channel name, the retention time, acquisition details, precursor observed mass, precursor charge states identified, the residual %precursor ions detected and the % coverage supplemented with a simplified Dotmap™ representation of confirmed monomers. Ordering this table by sequence coverage showed that two individual MS/MS conditions were able to produce 100% sequence coverage (charge state -9 at 10V and 15V collision energy). The % precursor drops from 91.62% at 10V to 46.81% at 15V. The precursor was not detected for this charge state when 20v was used, which indicates that over fragmentation has occurred at this energy.
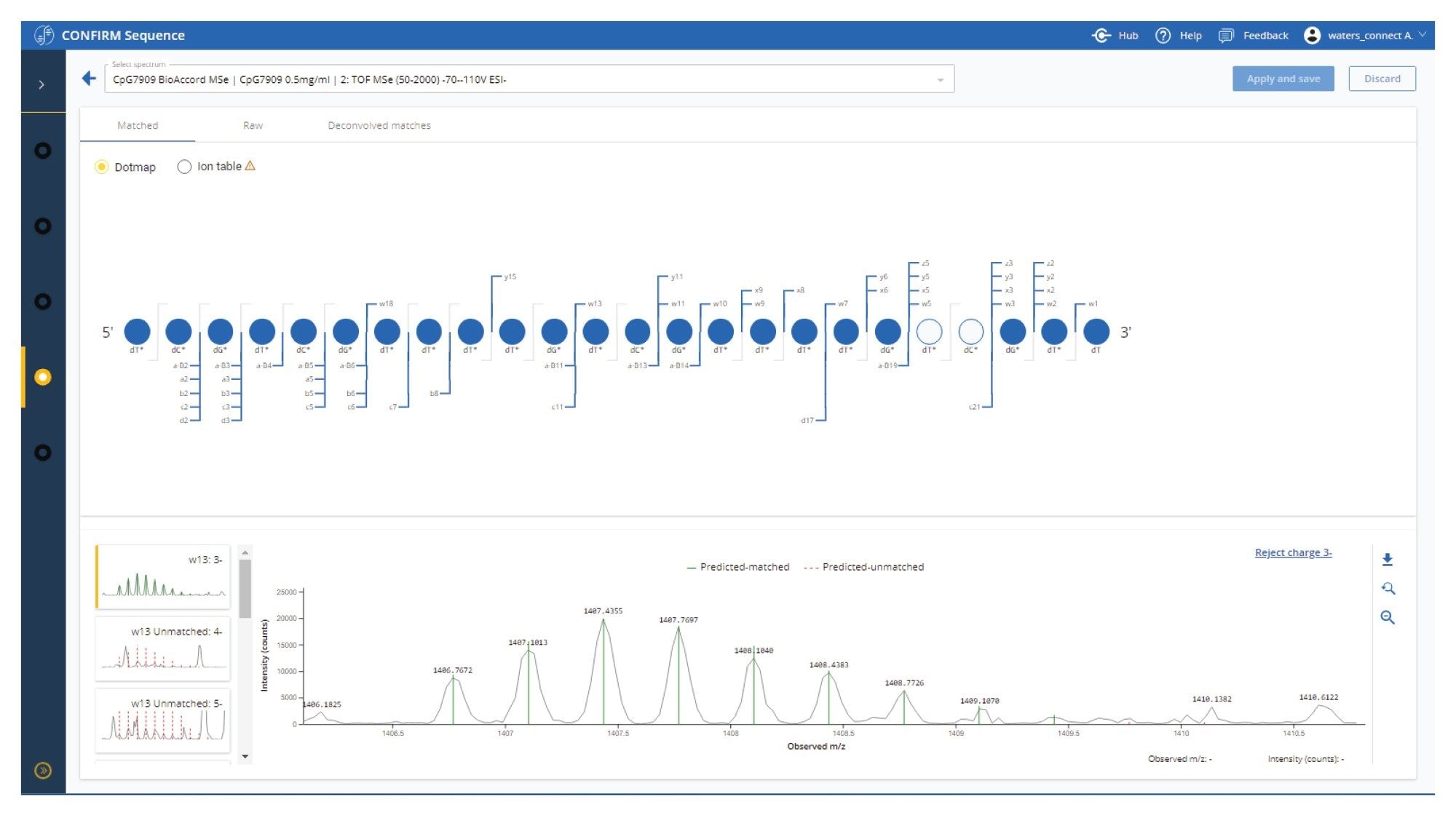
CpG7909, sequence dT*dC*dG* dT*dC*dG* dT*dT*dT* dT*dG*dT* dC*dG*dT* dT*dT*dT* dG*dT*dC* dG*dT*dT (d refers to a deoxyribonucleic acid, * refers to a phosphorothioate backbone modification).
Conclusion
The CONFIRM Sequence waters_connect™ application works well for both targeted MS/MS and data-independent fragmentation data.
Manual deconvolution and fragment assignment for synthetic oligonucleotide sequence confirmation is time consuming, more prone to user error, and highly dependent on user experience. In contrast, the automated CONFIRM Sequence workflow produced comparable results to manual workflows, but includes the benefits of automated fragment matching, matching directly on the raw data, improved processing speed, and consideration of isobaric ambiguities in the sequence coverage results.
The BioAccord extends the usability of the automated data processing for regulated manufacturing and quality labs, by providing a small footprint and cost-effective LC-MS system designed for non-MS experts on a compliance-ready platform. The CONFIRM Sequence App is capable of processing and annotating the DIA fragmentation data from BioAccord System and enabling high confidence assignments in an automated workflow. Good quality high energy data obtained from this TOF MS system makes it an attractive alternative to QTOF or other high-end mass spectrometry.
QTOF systems that possess greater fragmentation control and ultimate sensitivity for the initial characterization of a molecule may prove overkill for such routine sequence confirmation challenges. The utility of the CONFIRM Sequence App for supporting both workflows, enables a reduced training burden and for smoother transitions of molecules and methods from development to manufacturing and QC organizations.
References
- Sharma, V.K., Watts, J.K.: Oligonucleotide Therapeutics: Chemistry, Delivery, and Clinical Progress. http://dx.doi.org/10.4155/fmc.15.144. 7, 2221–2242 (2015). https://doi.org/10.4155/FMC.15.144
- Al Shaer, D., Al Musaimi, O., Albericio, F., de la Torre, B.G.: 2021 FDA TIDES (Peptides and Oligonucleotides) Harvest. Pharmaceuticals. 15, (2022). https://doi.org/10.3390/PH15020222
- Musaimi, O. Al, Shaer, D. Al, Albericio, F., de la Torre, B.G.: 2020 FDA TIDES (Peptides and Oligonucleotides) Harvest. Pharmaceuticals. 14, 1–14 (2021). https://doi.org/10.3390/PH14020145
- Dhuri, K., Bechtold, C., Quijano, E., Pham, H., Gupta, A., Vikram, A., Bahal, R.: Antisense Oligonucleotides: An Emerging Area in Drug Discovery and Development. J Clin Med. 9, 1–24 (2020). https://doi.org/10.3390/JCM9062004
- Rossi, J.J., Rossi, D.J.: siRNA Drugs: Here to Stay. Molecular Therapy. 29, 431–432 (2021). https://doi.org/10.1016/J.YMTHE.2021.01.015
- McLuckey, S.A., Habibi-Goudarzi, S.: Ion Trap Tandem Mass Spectrometry Applied to Small Multiply Charged Oligonucleotides with a Modified Base. J Am Soc Mass Spectrom. 5, 740–747 (1994). https://doi.org/10.1016/1044-0305(94)80006-5
- Mcluckey, S.A., Van Berkel, G.J., Glish, G.L.: Tandem Mass Spectrometry of Small, Multiply Charged Oligonucleotides. J Am Soc Mass Spectrom. 3, 60–70 (1992). https://doi.org/10.1016/1044-0305(92)85019-G
- Abdullah, A.M., Sommers, C., Hawes, J., Rodriguez, J.D., Yang, K.: Tandem Mass Spectrometric Sequence Characterization of Synthetic Thymidine-Rich Oligonucleotides. Journal of Mass Spectrometry. 57, (2022). https://doi.org/10.1002/JMS.4819
- Wu, J., McLuckey, S.A.: Gas-Phase Fragmentation of Oligonucleotide Ions. Int J Mass Spectrom. 237, 197–241 (2004). https://doi.org/10.1016/J.IJMS.2004.06.014
- Gawlig, C., Rühl, M.: Investigation of the Influence of Charge State and Collision Energy on Oligonucleotide Fragmentation by Tandem Mass Spectrometry. Molecules 2023, Vol. 28, Page 1169. 28, 1169 (2023). https://doi.org/10.3390/MOLECULES28031169
- Cooper, C.L., Davis, H.L., Morris, M.L., Efler, S.M., Al Adhami, M., Krieg, A.M., Cameron, D.W., Heathcote, J.: CPG 7909, An Immunostimulatory TLR9 Agonist Oligodeoxynucleotide, as Adjuvant to Engerix-B® HBV Vaccine in Healthy Adults: A double-blind phase I/II study. J Clin Immunol. 24, 693–701 (2004). https://doi.org/10.1007/S10875-004-6244-3/METRICS
Acknowledgements
Alexander Litz at BioSpring GmbH for assisting with data generation.
waters_connect, Vion, BioAccord UPLC, ACQUITY and RDa are trademarks of Waters Technologies Corporation.
We thank Hüseyin Aygün for enabling the presented study at BioSpring GmbH.
Michael Rühl and Güngör Hanci are employees of BioSpring GmbH, Alt-Fechenheim 34, 60386 Frankfurt, Germany.
720008079, November 2023