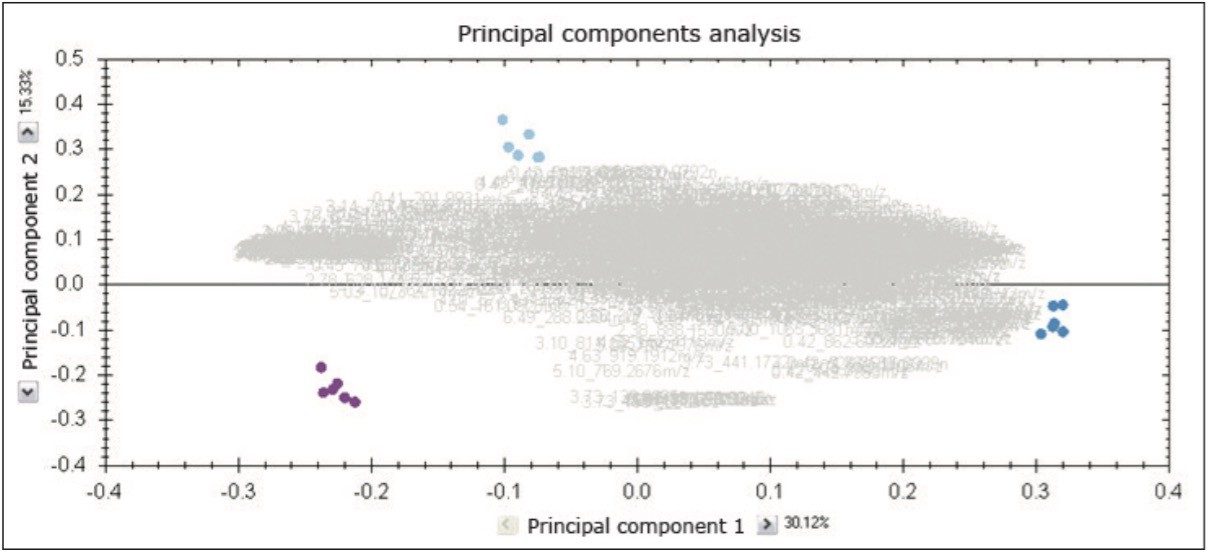
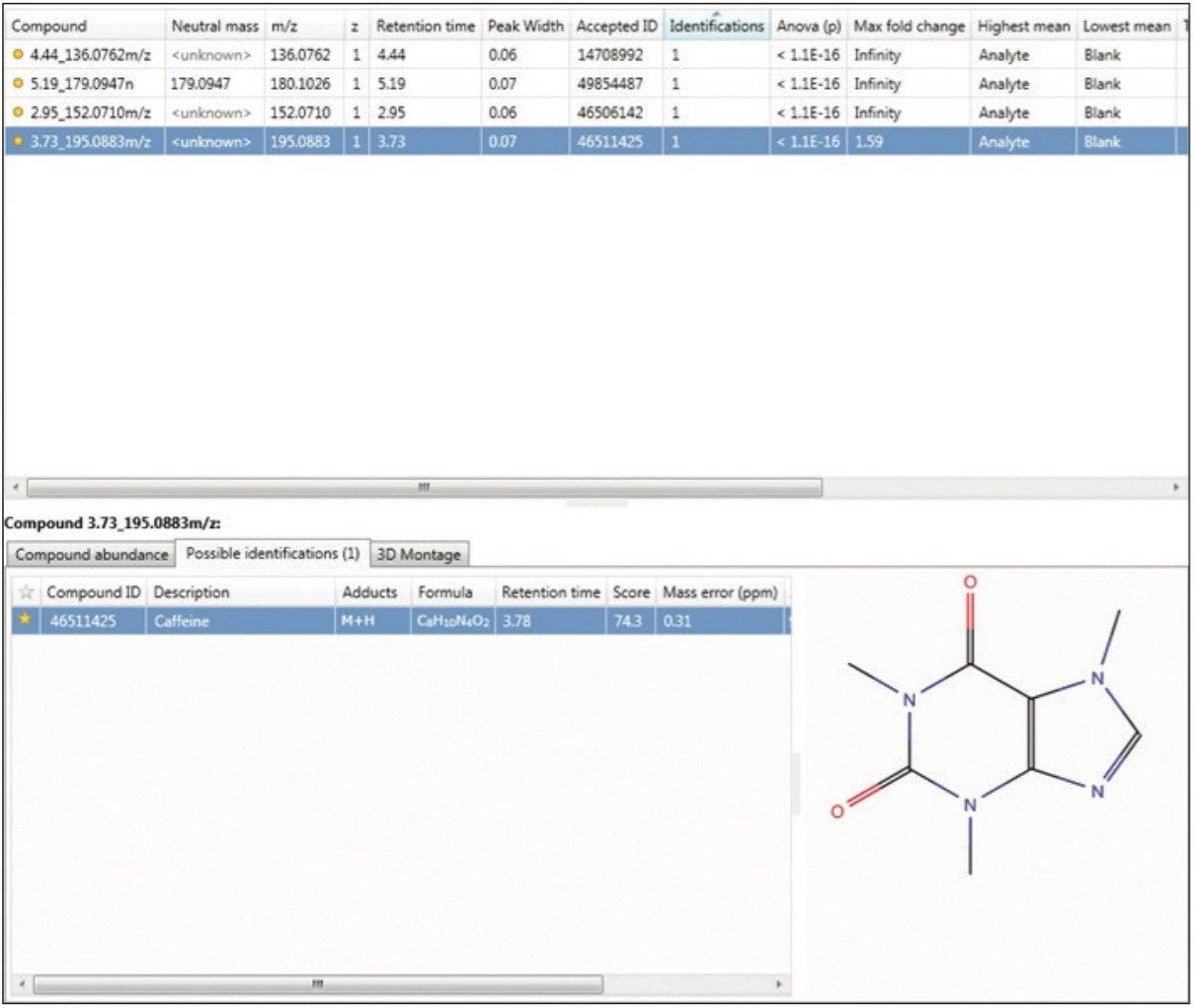
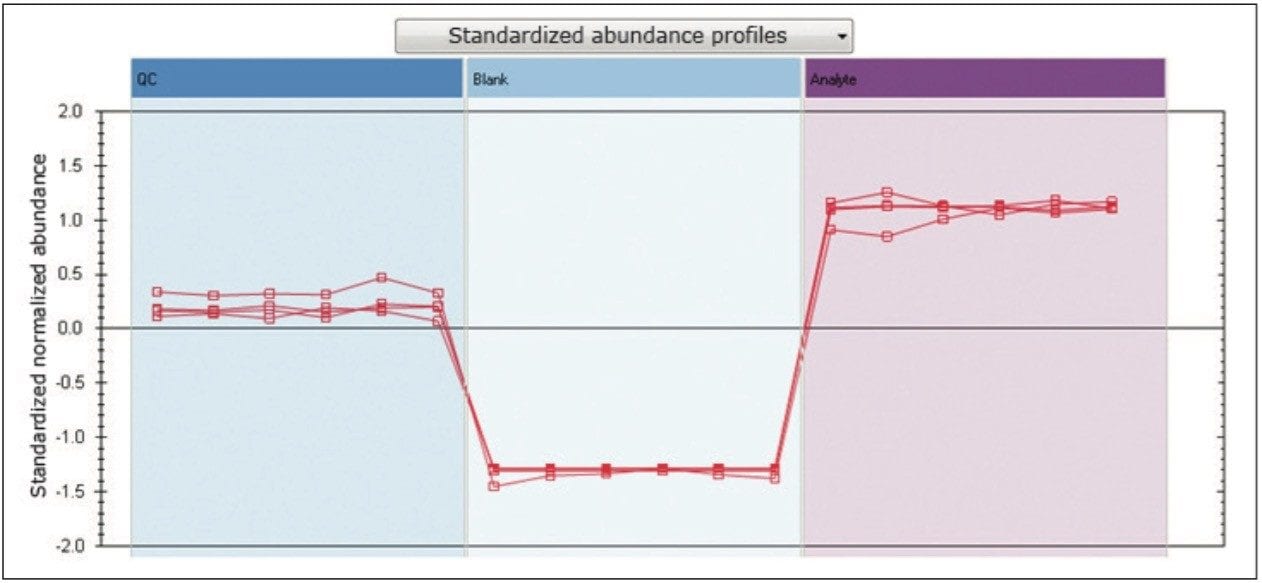
Moreover, TOIML enables the scientist to link the analysis results with other omics data or provide input into separate statistical software packages such as EZinfo. Results from downstream bioinformatics (i.e., Umetrics software) can be imported back into an analyzed experiment, combining all compound data into a single table to review or share.
For the proteomics experiment, three replicates of two 10-ng E.coli samples differentially spiked with bovine serum albumin (BSA), alcohol dehydrogenase (ADH), enolase, and glycogen phosphorylase B were analyzed. The injected on-column amounts for the spiked protein in the first sample (Mixture 1) were one femtomoles each and 8, 1, 2, and 0.5 femtomoles for the second sample (Mixture 2), respectively. The nominal expected ratios (Mixture 2:Mixture 1) were therefore 8:1, 1:1, 2:1, and 0.5:1. Here, the peptides were separated and analyzed using a nanoACQUITY UPLC System coupled to a Xevo G2-S QTof and operated in LC-MSE acquisition mode. Processing, searching, and quantification were conducted with TransOmics for Proteomics (TOIP) using a species specific database to which sequence information of the spiked proteins was appended.
The TOIP procedure involves the following steps:
1. Importing the raw MSE continuum data set (three technical replicates per sample)
2. Peak alignment to correct retention time drift between analytical runs
3. Chromatographic peak normalization to allow comparison across different sample runs
4. Chromatographic peak detection (peak picking)
5. Protein and peptide identification utilizing integrated database search algorithms
6. Multivariate statistical analysis
7. Absolute and relative quantitation
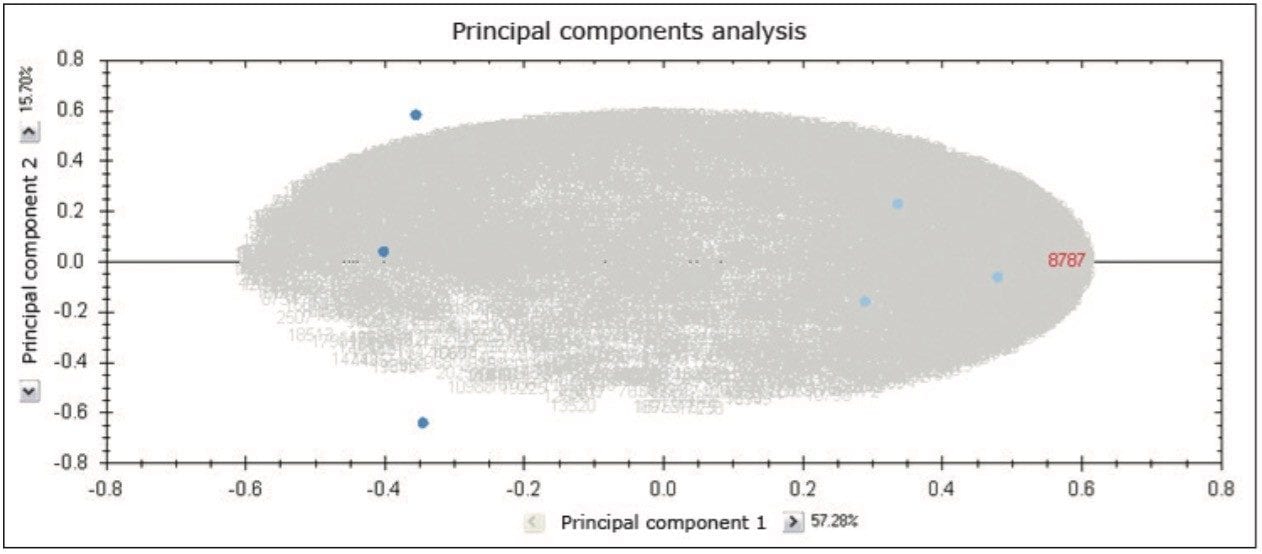
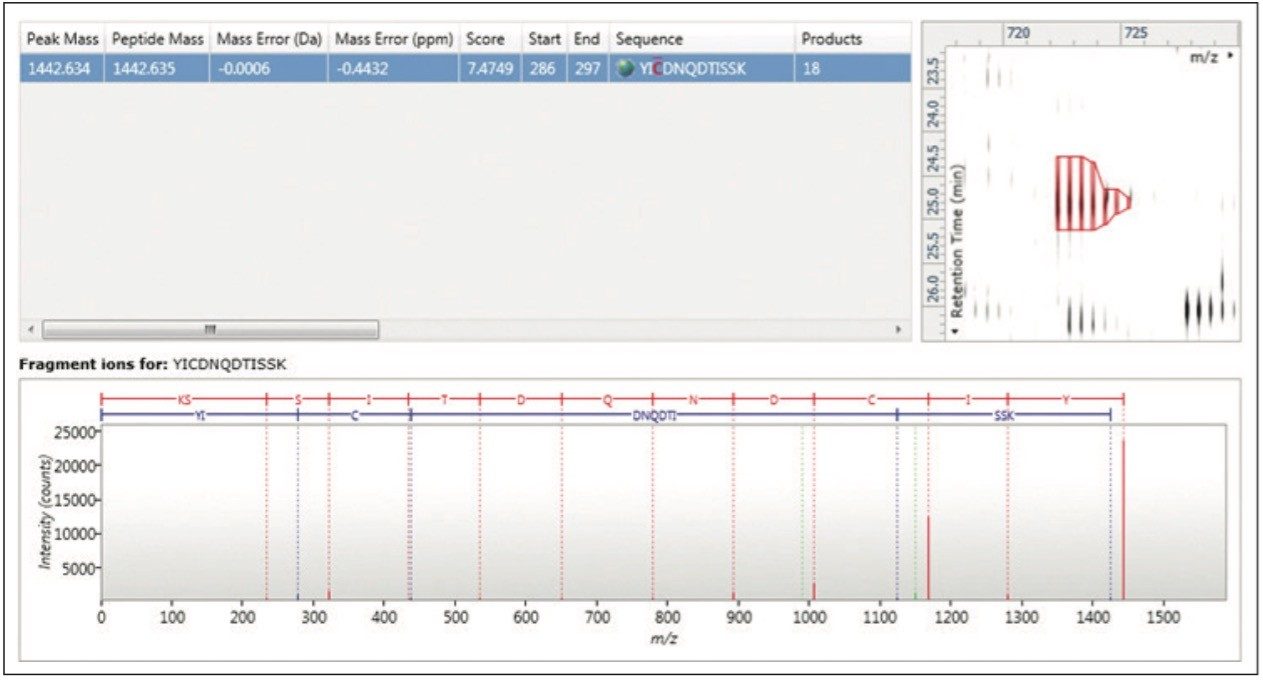
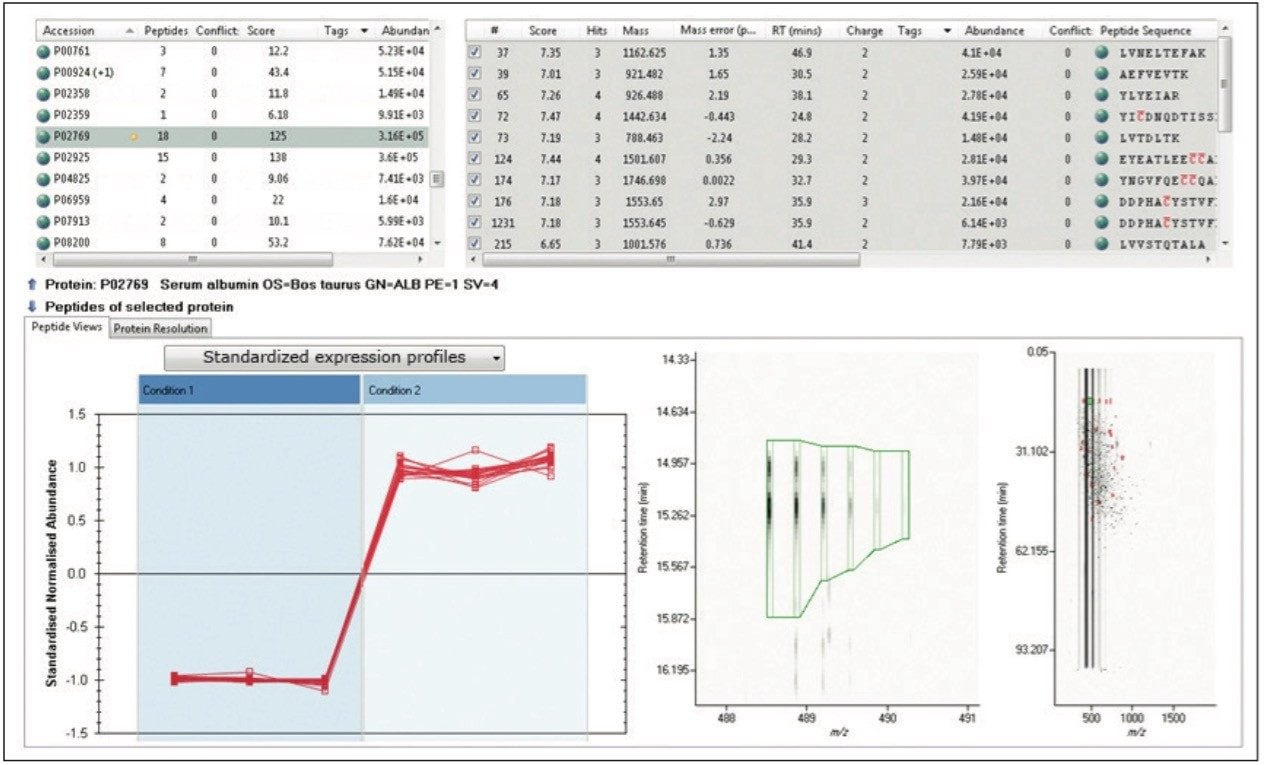
TOIP offers the same multivariate analysis tools as TOIML. Figure 4 illustrates an example of PCA of the detected features, i.e. charge state groups. Primary clustering at the technical replicate level can be readily observed. A qualitative peptide identification result for one of the spiked protein digest is shown in Figure 5, and the normalized expression profiles of all peptides identified to this protein are shown in Figure 6. The latter is demonstrative for the type of quantitation precision that can be obtained by means of label-free MS studies using and LC-MSE- based acquisition strategy.