Initial screening
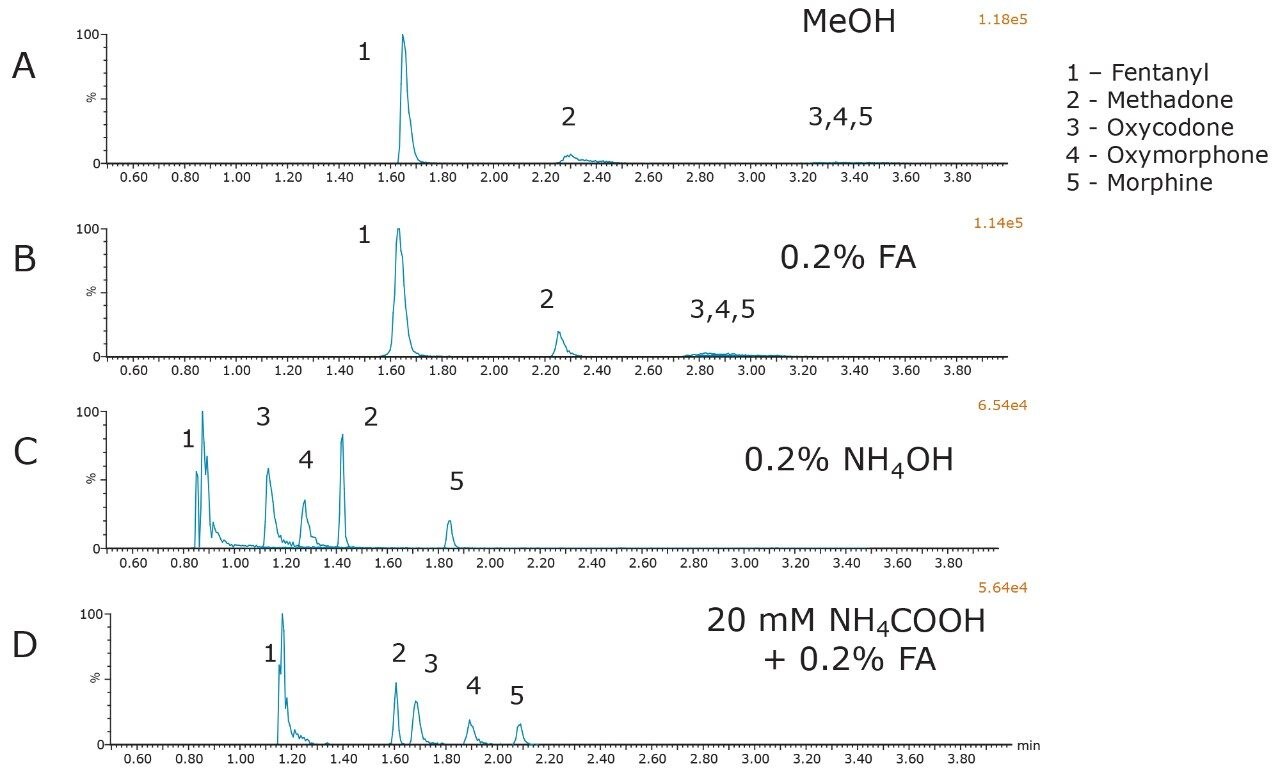
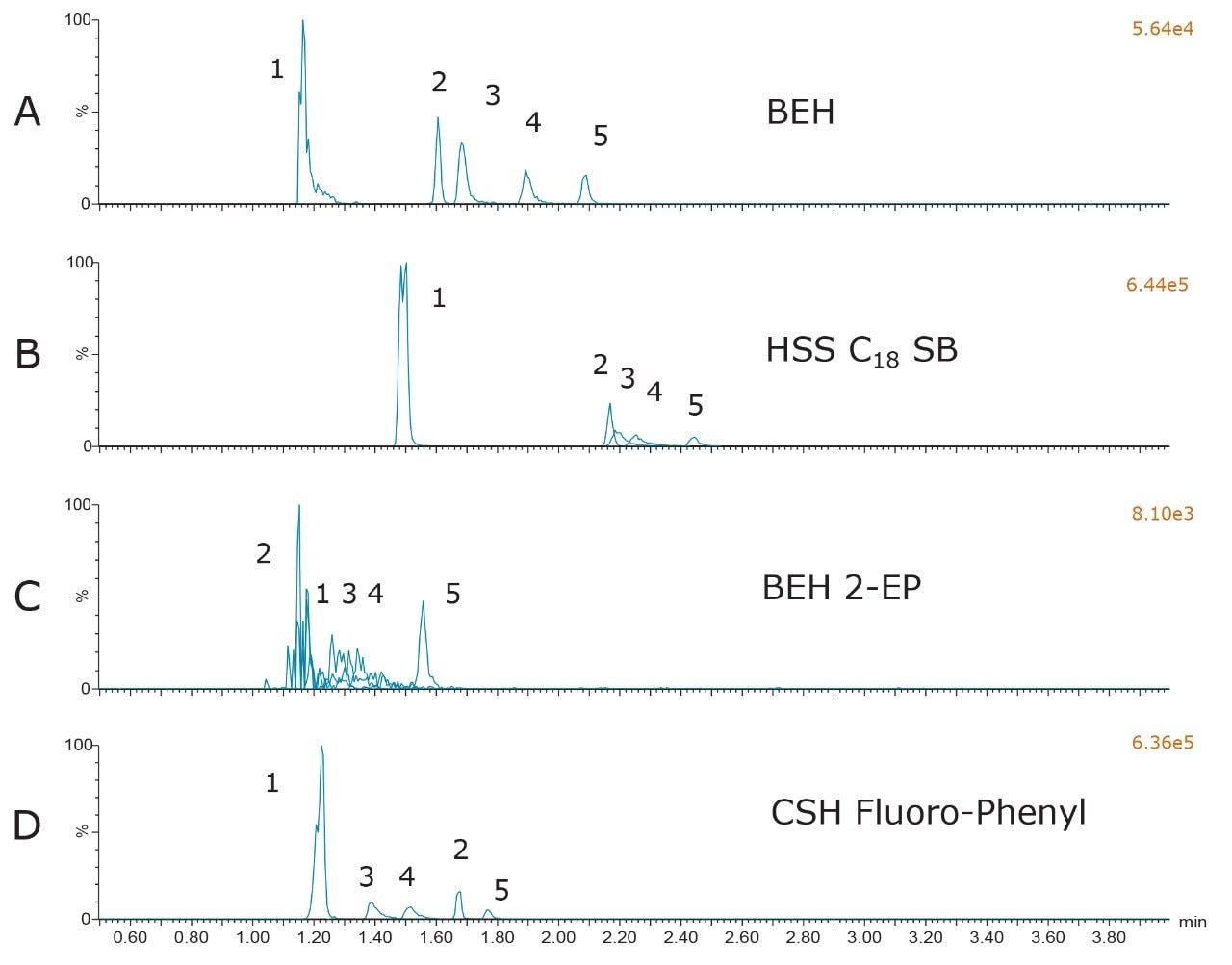
In order to determine the optimal starting conditions for the analysis of opioid drugs using UPC2, a series of screening runs was performed using a four-position column manager to evaluate four different column chemistries and four different mobile phase additives. The columns used were Waters UPC2 BEH, BEH 2-EP, CSH Fluoro-Phenyl, and HSS C18 SB. Methanol was used for the B mobile phase with the following additives: None (MeOH only), 0.2% formic acid, 0.2% NH4OH, or 0.2% formic acid + 20 mM NH4COOH. The initial screening gradient started at 5% B and increased to 75% B over 4 min. The flow was returned to 5% B over 1 min, and held at the initial conditions for 1.4 min to re-equilibrate the column. Flow rates for each column were set to keep the back pressure below the system limit of 6000 psi and were 1.5 mL/min for the BEH and 2-EP columns and 1.0 mL/min for the HSS and PFP CSH Fluoro-Phenyl columns. Initial screenings were performed with a limited group of compounds including fentanyl, morphine, oxymorphone, oxycodone, and methadone. These represent a range of polarities chosen to simplify the initial screening process.
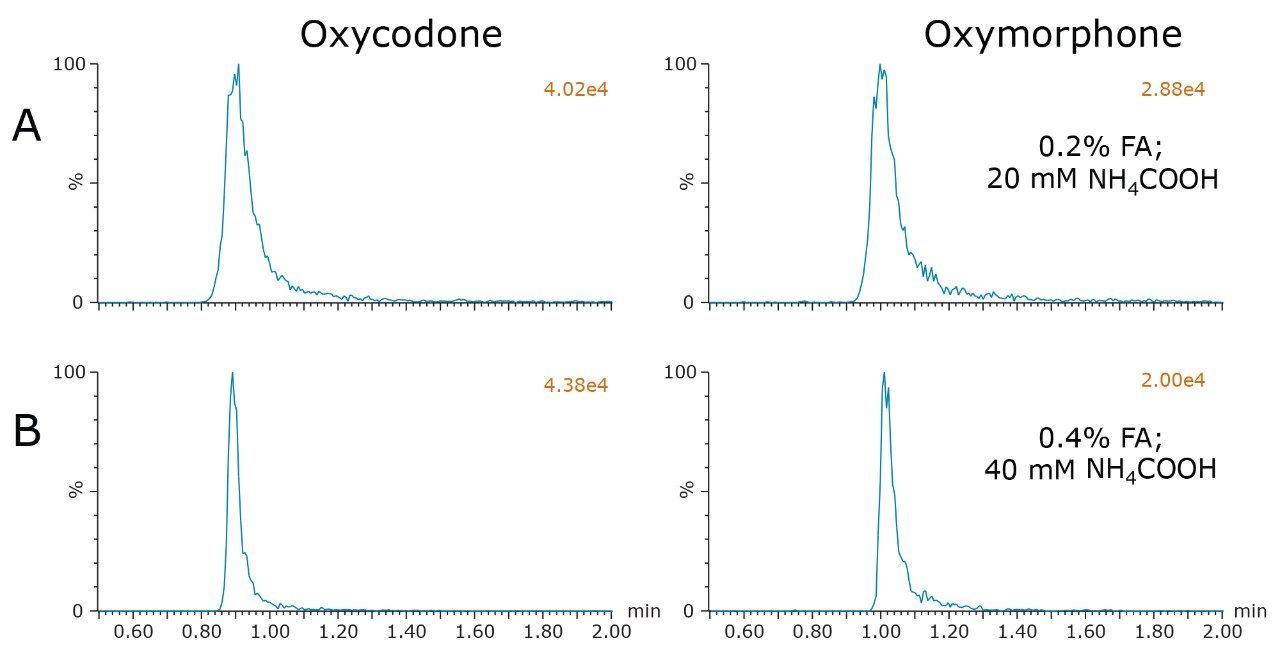
The initial evaluation of MPB additives on the BEH column is shown in Figure 1. All compounds were at a concentration of 500 ng/mL. It is quite apparent from this figure that both pure methanol and the addition of 0.2% formic acid produced broad peak shapes with low intensity in MS for oxycodone, oxymorphone, and morphine. In contrast, the addition of either 0.2% NH4OH or 0.2% formic acid + 20 mM NH4COOH resulted in acceptable peaks for most of the compounds used in this initial screen. This is consistent with previous reports of alkaline conditions being used to achieve good chromatographic performance for bases in general, and opiates in particular under SFC conditions.1,2 Closer evaluation of the bottom two chromatograms revealed that the buffered additive resulted in greater retention and improved peak shape compared to the addition of concentrated ammonia only