UPLC separation
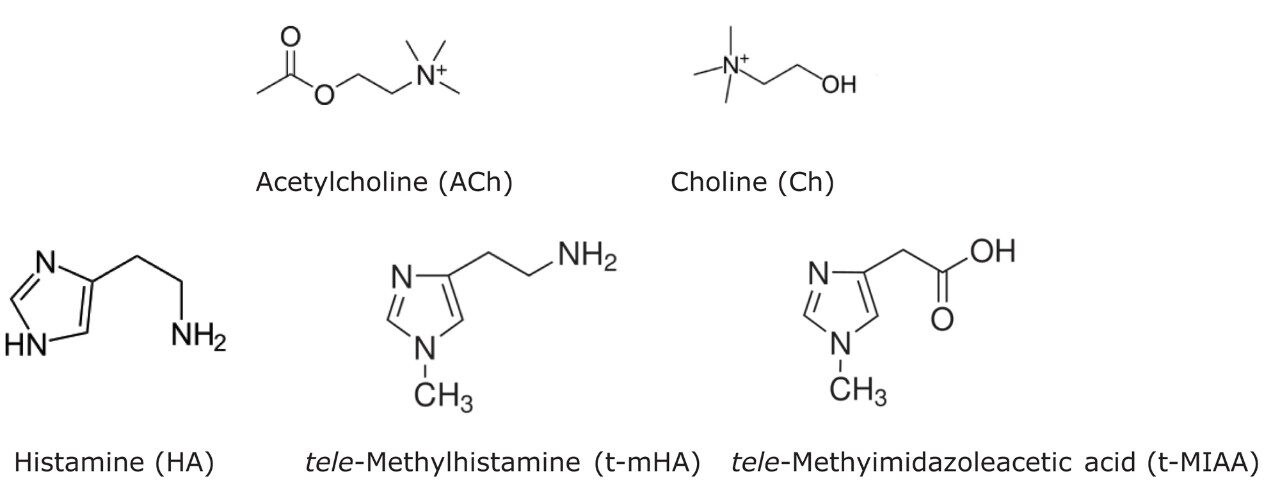
ACh, Ch, HA, t-mHA, and t-MIAA are highly polar, low molecular weight compounds present at relatively low concentrations, making them difficult to detect by standard reversed-phase (RP) chromatography. Additionally, quantitative analysis of these endogenous analytes in biological matrix is often difficult due to matrix-induced interferences and ion suppression from co-elution with related molecules, such as salts or other low molecular weight endogenous components.9
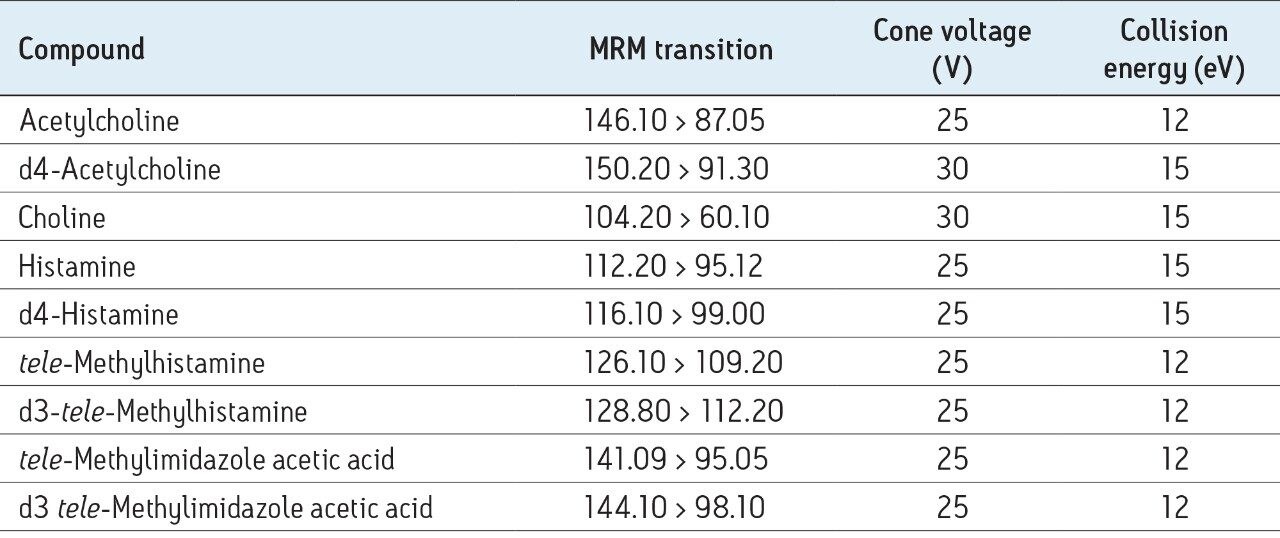
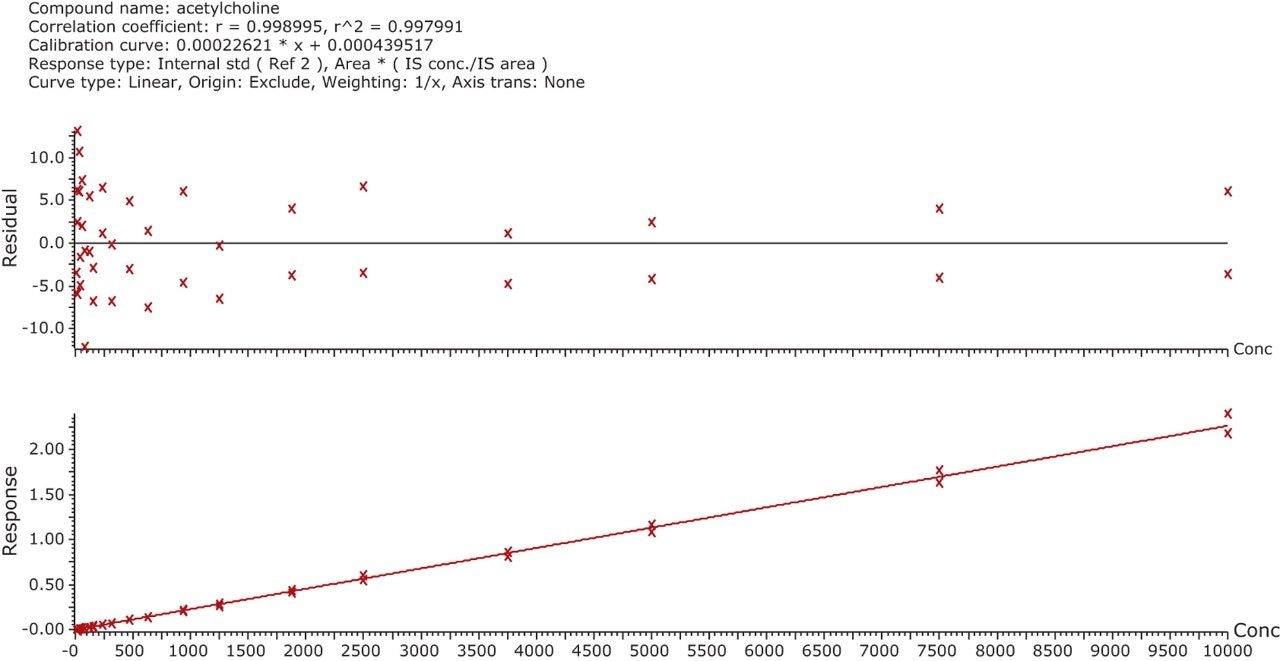
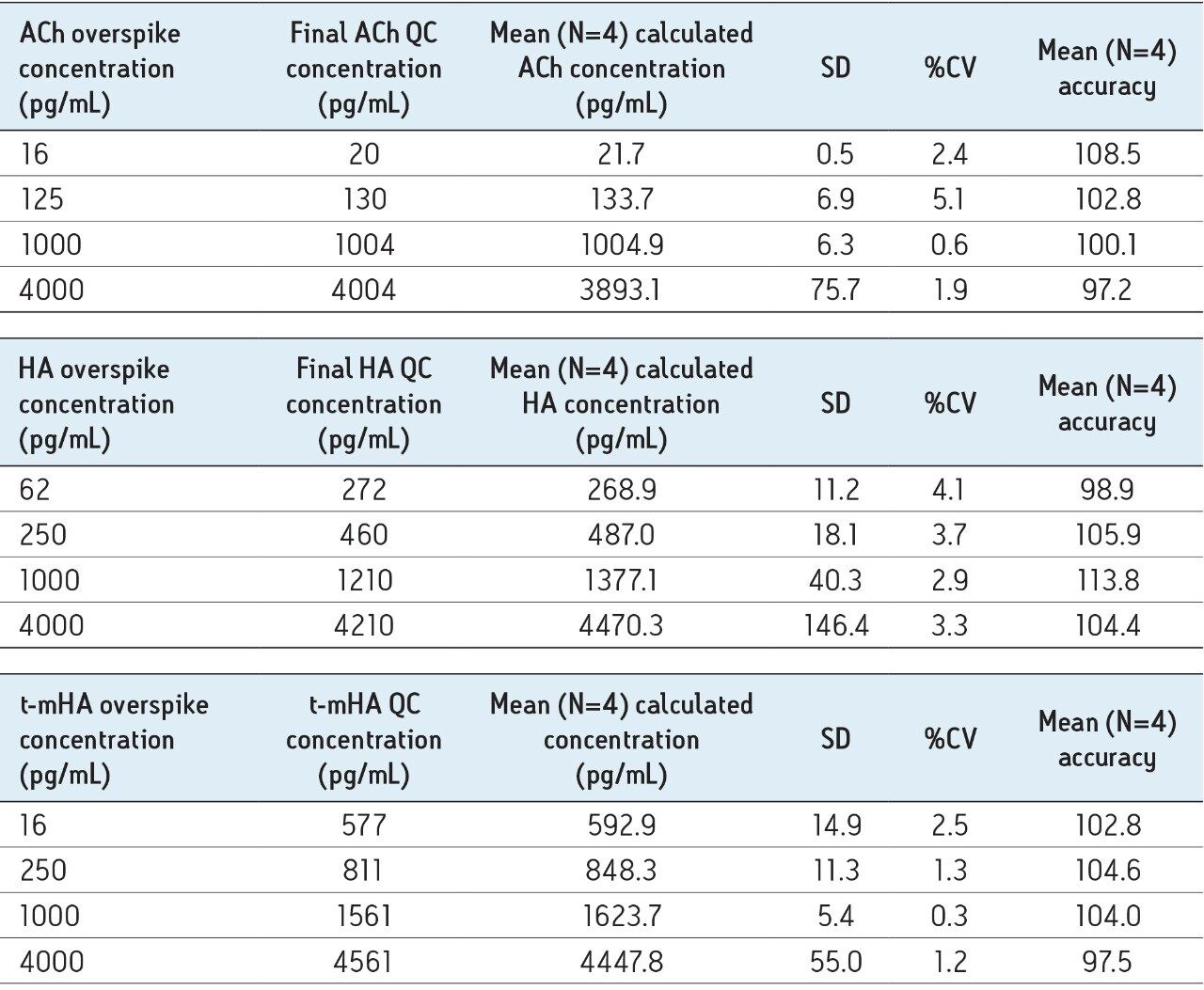
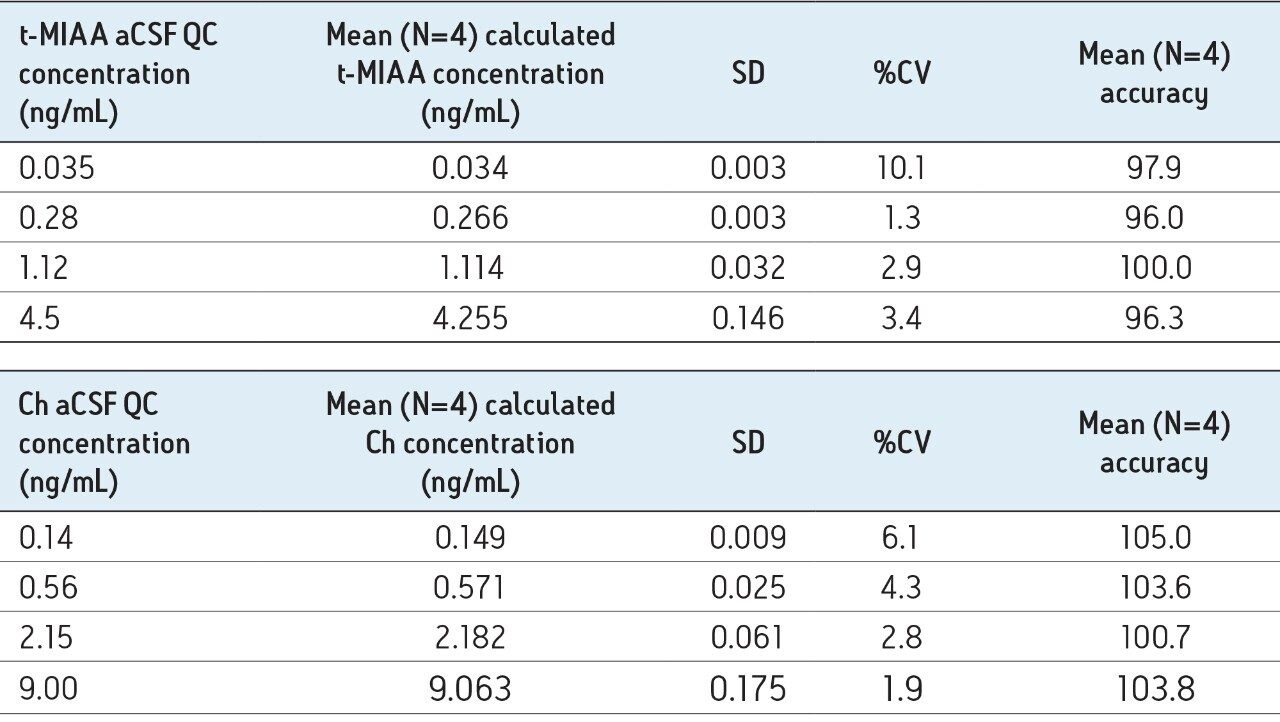
In this application, chromatographic separation of ACh, HA, and their respective metabolites was achieved utilizing a sub-2-µm CORTECS UPLC HILIC Column on an ACQUITY UPLC System. The optimum ammonium formate concentration (mobile phase A) was determined to be 100 mM, pH 3, while mobile phase B consisted of acetonitrile. An optimum flow rate of 0.5 mL/min using a gradient from 90% to 30% B was employed with a cycle time of 2.5 minutes at a column temperature of 45 °C.
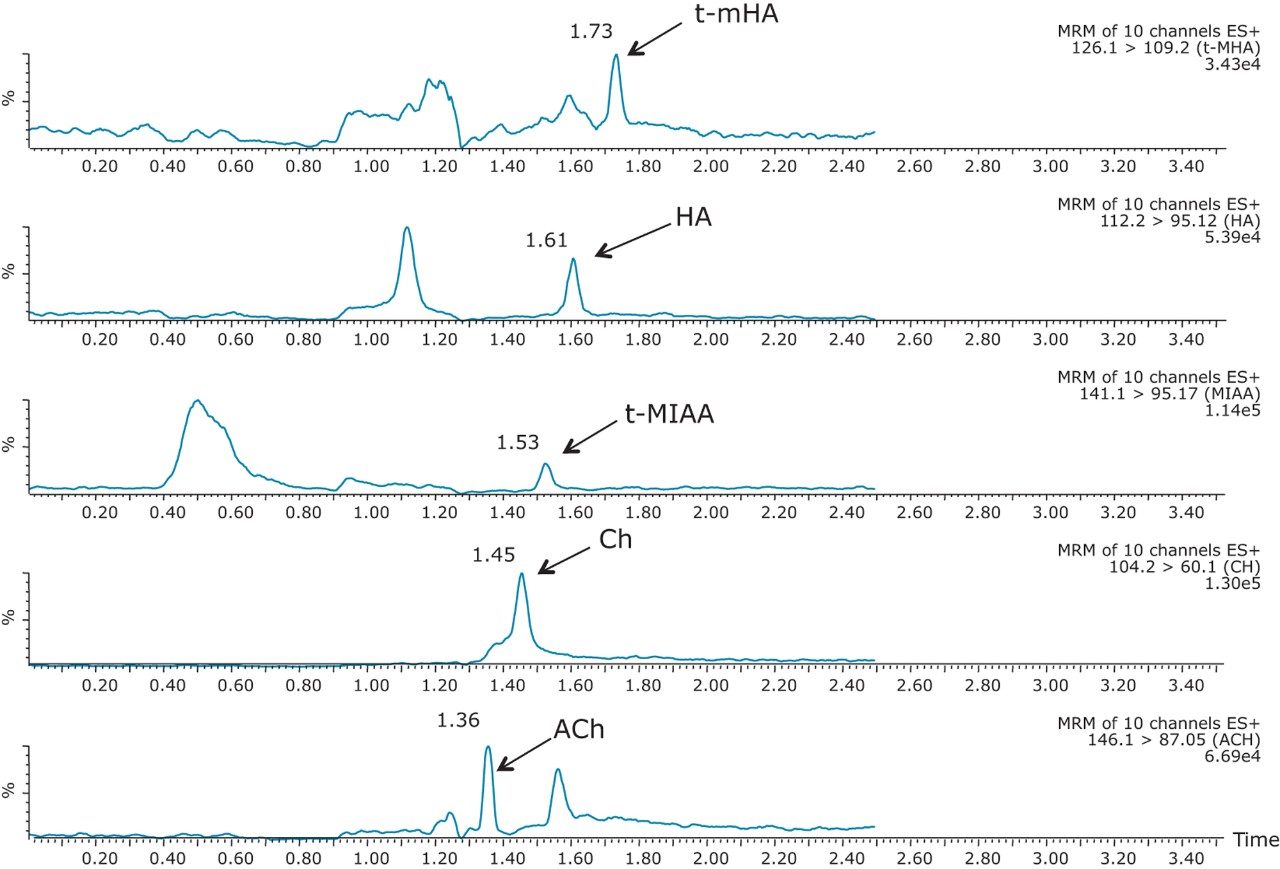
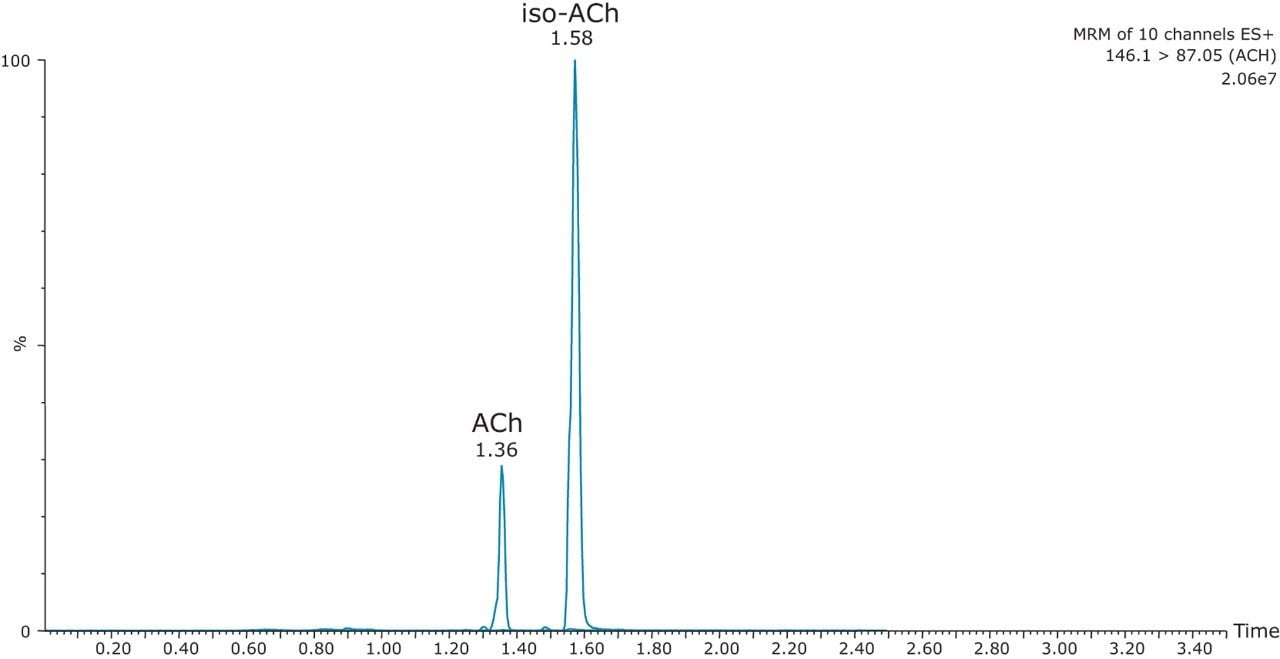
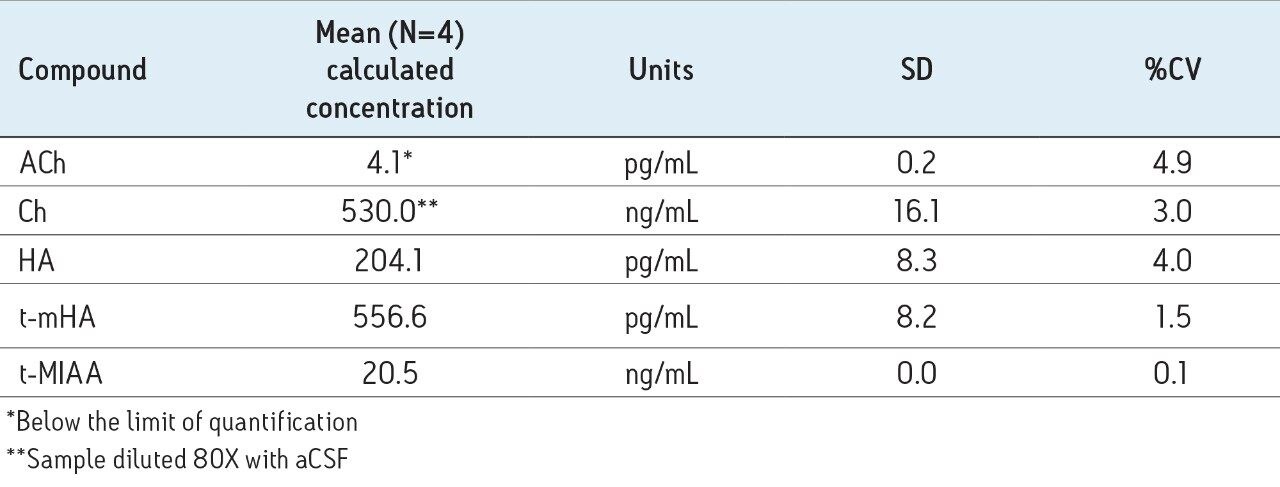
The mobile-phase additive concentration greatly influenced the specificity, sensitivity, and the ability to chromatographically resolve the analytes of interest from both mobile phase and endogenous matrix interferences. The higher buffer concentration of 100 mM (rather than 20 to 50 mM) greatly improved peak widths for the later eluting analytes. Specifically, peak tailing of t-MIAA was significantly reduced with the use of a high mM concentration of ammonium formate. Chromatographic retention and performance for the lower limit of quantification control (LLQC) samples of ACh, Ch, and their respective metabolites is shown in Figure 2. ACh, HA, their metabolites, as well as an isobar of ACh, Iso-ACh (3-carboxypropyl) trimethylammonium), eluted between 1.35 and 1.75 minutes, with peak widths less than 3.7 seconds at base.