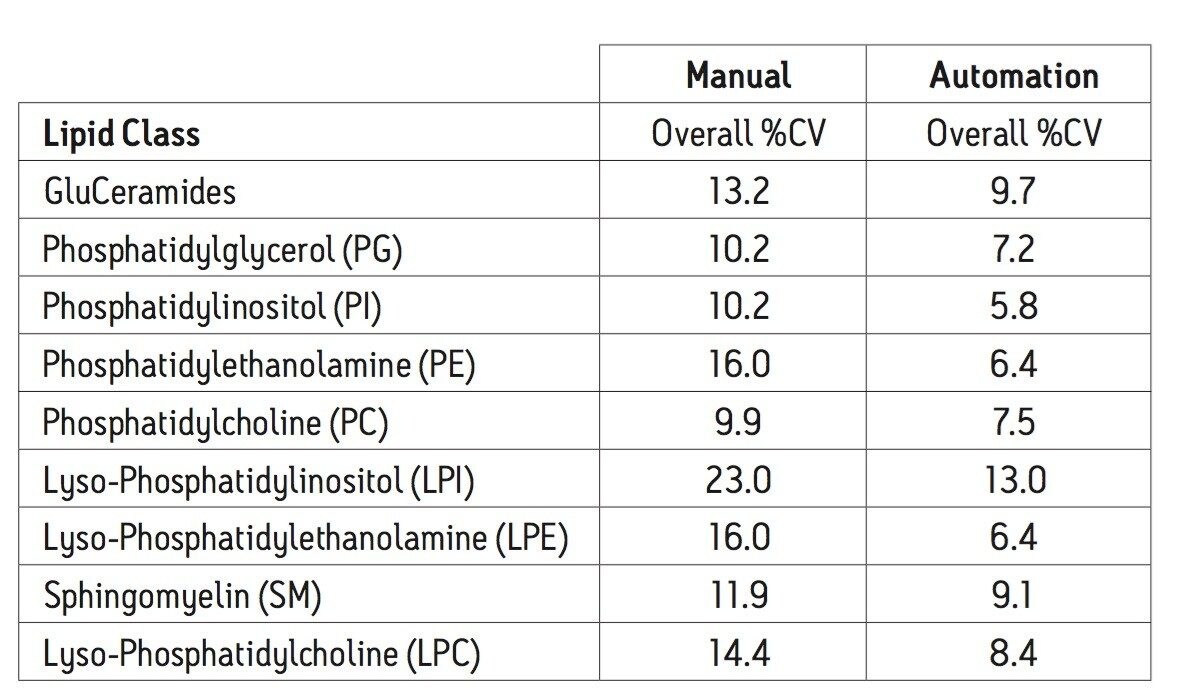
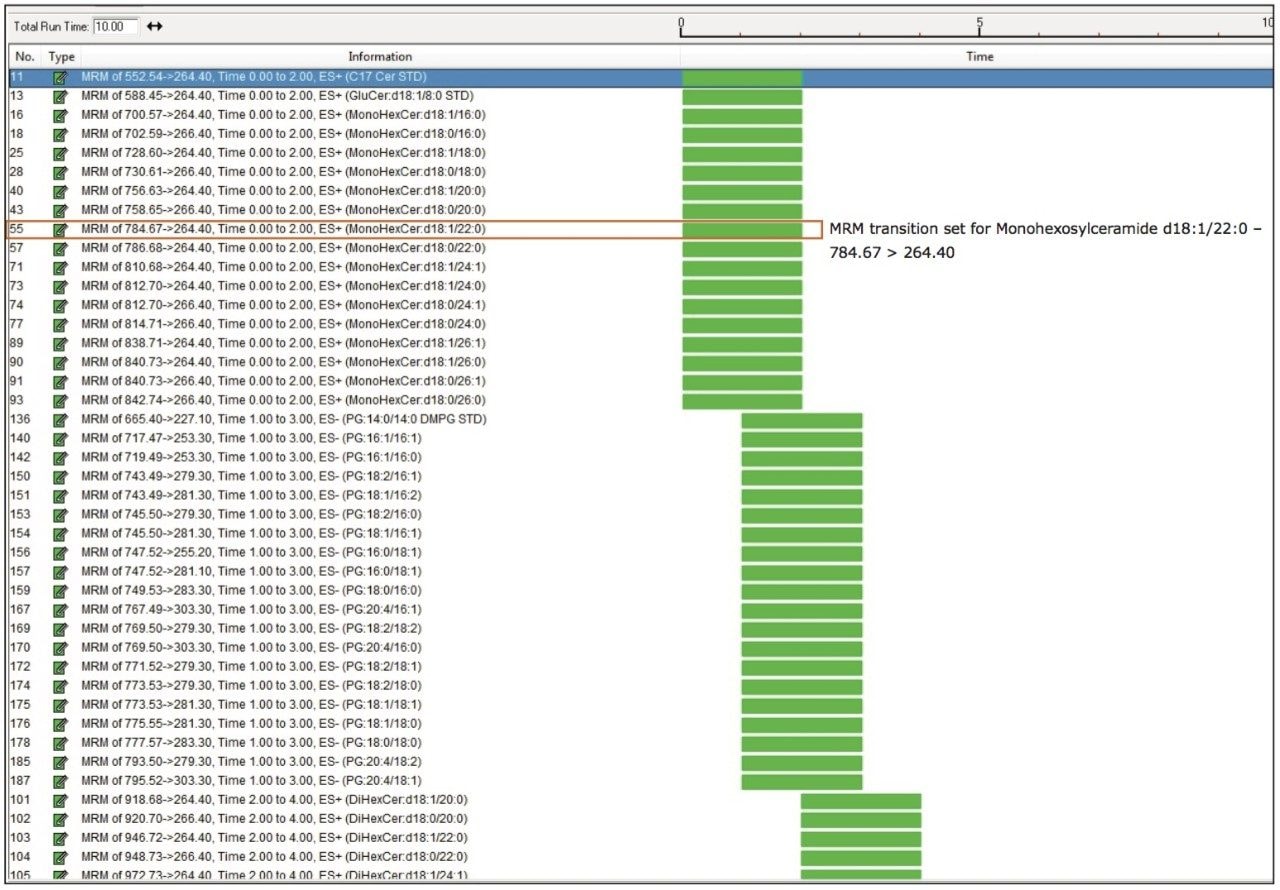
Lipids play many important roles in maintaining homeostasis of living organisms. Lipidomics analyses could further our understanding of mechanisms of disease, including the identification of biomarkers and potential drug targets.
Biofluids such as plasma are typically complex, with large lipid diversity across many orders of concentration. These, together with the chemical complexity of lipids, present demanding analytical challenges ranging from the sample preparation stage to the analytical techniques used to identify and quantify key lipids. Today, many variations of the Bligh and Dyer method are used for total lipid extraction and purification, with equal amounts used for mass spectrometric analysis.
Recent advances in lipidomics have made use of developments in chemistries and instrumentation, most notably the use of off-line enrichment or solid-phase sample preparation products1,2 and the coupling of UltraPerformance LC with mass spectrometry. However, there is little standardization across platforms and workflows for a complete analysis.
Presented here is a tandem quadrupole-based phospholipid analysis workflow from extraction to separation, identification and quantification of the phospholipids from a single vendor. These commercially available products packaged as a complete solution are provided to ease the strains and increase the productivity of laboratories undertaking longitudinal studies spanning hundreds of lipids over thousands of samples.