Xevo QTof: Sub-Femtomol Detection and Label-Free Quantification of Protein Digests
Abstract
Low abundance proteins are often of biological interest and as such, sensitivity and low limit of quantification are key parameters in modern proteomics experiments.
The optimized time-of-flight (Tof) geometry of Waters® Xevo™ QTof MS System provides high sensitivity, resulting in the routine detection of attomol levels of tryptically digested proteins.
In this technical note, we demonstrate accurate label-free protein quantification at high mass accuracy (3.5 ppm) on attomol levels of protein injected.
Benefits
Introduction
The use of LC/MSE to qualitatively and quantitatively characterize enzymatic protein digests is now standard and widespread throughout many proteomics laboratories. This method has proven to be highly efficient when dealing with protein mixtures of varying complexity.
LC/MSE data were acquired on a Xevo QTof Mass Spectrometer that incorporates a new high field pusher device and dual stage reflectron. This arrangement enables a resolution of greater than 10,000 FWHM to be achieved with a flight tube 35-cm in length. Additionally, a shorter flight tube allows for a higher pusher frequency, therefore, improving the oa-ToF duty-cycle and increasing sensitivity.
Experimental
Samples consisted of the Waters MassPREP™ Digestion Standards Protein Expression Mix 1 and 2 (p/n: 186002865 and p/n: 1860028656). Mix 1 contains alcohol dehydrogenase (P00330), enolase (P00924), bovine serum albumin (P02769), and phosphorylase-B (P00489) in equal amounts. In Mix 2, ADH is at a ratio of 1:1, enolase is up-regulated 2-fold, BSA is up-regulated 8-fold, and phosphorylase B down–regulated 2-fold. A dilution was made such that 500 attomol of each protein (based on Mix 1 and ADH of Mix 2) was injected onto the system.
The Xevo QTof was operated in LC/MSE mode over the m/z range 50 to 1900 in nanoelectrospray mode. The capillary, sample cone, extraction cone and collision energy were 3.8 kV, 24.0 V, 2.0 V, and 6.0 V respectively. During the elevated energy scan, the collision energy is ramped from 15 V to 40 V. Lock Mass correction was performed with Glu-fibrinopeptide B m/z 785.8426. Data was collected using MassLynx™ Software v.4.1 and processed using ProteinLynx™ Global Server 2.4.
The nanoACQUITY UPLC® System was operated at a flow rate of 300 nL/min and a system pressure of 3500 psi. The gradient was a linear 0% to 40% acetonitrile (0.1% v/v formic acid) over 40 minutes, with a a 75 µm x 100 mm C18 BEH 1.7 µm analytical column. Samples were loaded in the direct loading mode.

Figure 2 shows the PLGS 2.4 processed MSE data file representing a 500 amol equimolar load of each protein in Mix 1. RMS mass errors of better than 3.5 ppm were achieved on all identified precursor ions of the four proteins. The aforementioned mass accuracies are independent of m/z range and scan time used, and are routinely achievable on a 35 cm flight tube. The ability to routinely obtain sub-5 ppm RMS mass accuracies on the precursor, in combination with high-mass accuracy fragment ion information, generated in the elevated LC/MSE scan, provides a high-degree of specificity and significantly reduces false positive identification rates.
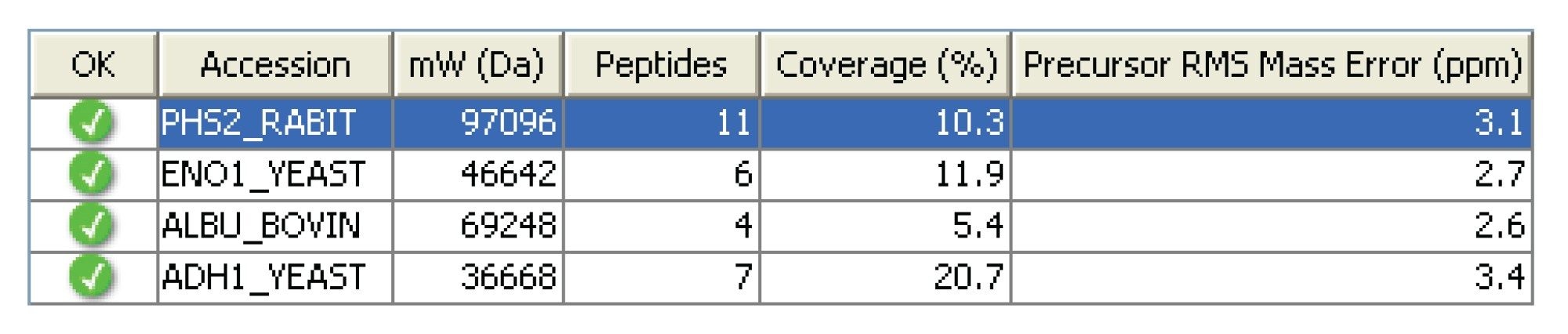
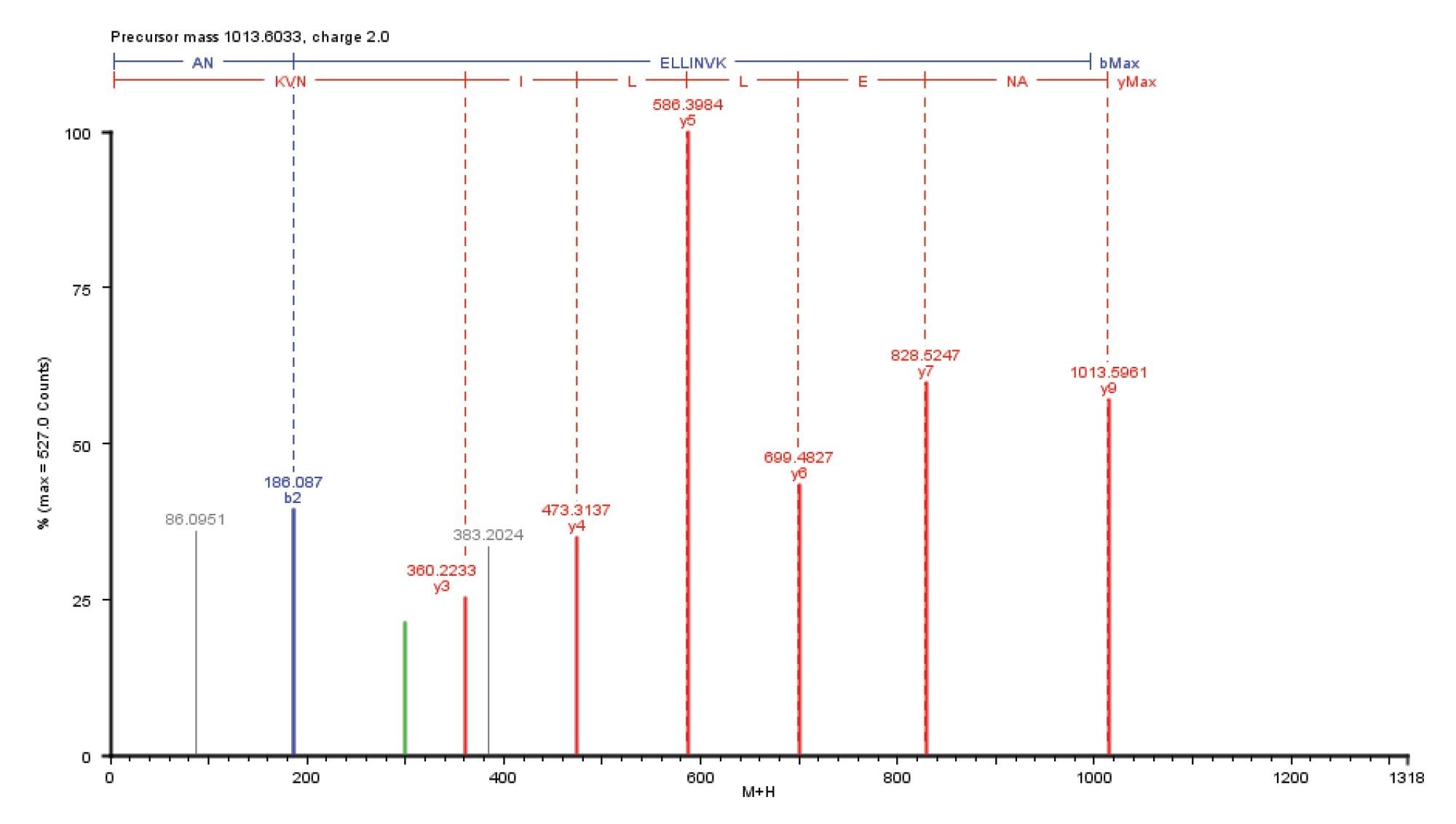
Figure 3 is an example of the PLGS2.4 processed MSE elevated energy data for the doubly charged alcohol dehydrogenase tryptic peptide ion m/z 507.8. Full amino acid sequence coverage (nine residues) of this peptide was achieved on a 500 amol loading level.
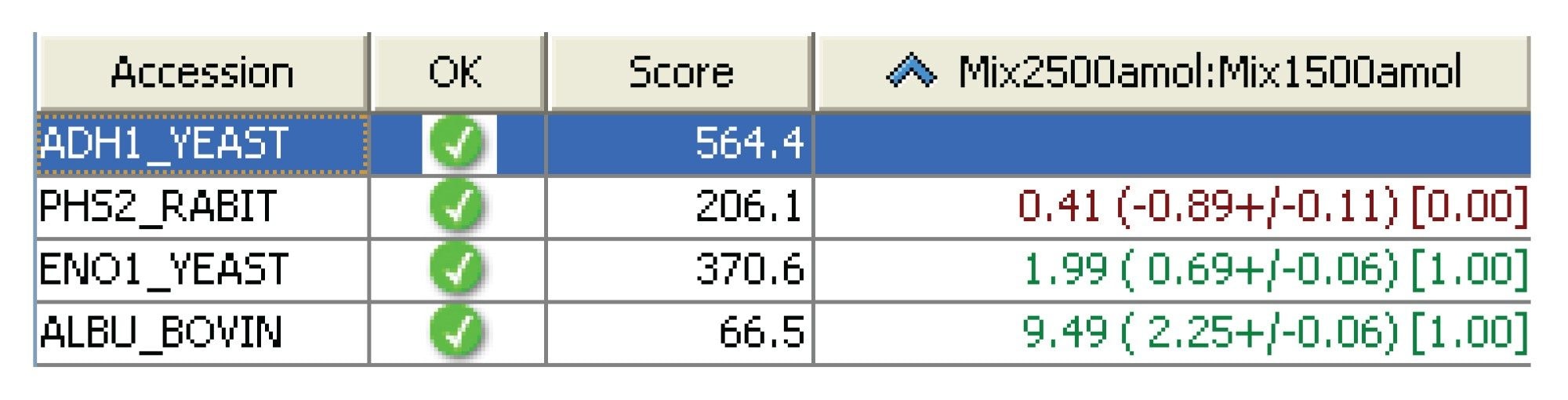
Utilizing an internal protein standard of known concentration, one can perform relative and absolute protein quantitation, using the top three best-ionizing peptides. In this case, alcohol dehydrogenase was used as the internal standard, since in both Mix 1 and Mix 2 alcohol dehydrogenase were present at 500 amol and relative protein quantification was carried out between Mix 1 and Mix 2 using all of the available matching peptides. The method of quantification used here was a label-free approach that does not require any chemical modification of the protein digest, which particularly at low levels may result in sample losses.
Conclusion
- The results presented here demonstrate the inherent sensitivity of the nano ACQUITY UPLC System and the Xevo QTof MS combination operated in LC/MSE mode, which allowed for the reliable and routine analysis of sub-femtomol levels of protein digest
- RMS mass accuracy below 3.5 ppm were obtained on all four protein standards in the MassPREP digestion standard Mix 1.
- Accurate relative quantification of the four protein mixture has been demonstrated using 500 amol of alcohol dehydrogenase as an internal protein standard
720003340, February 2010