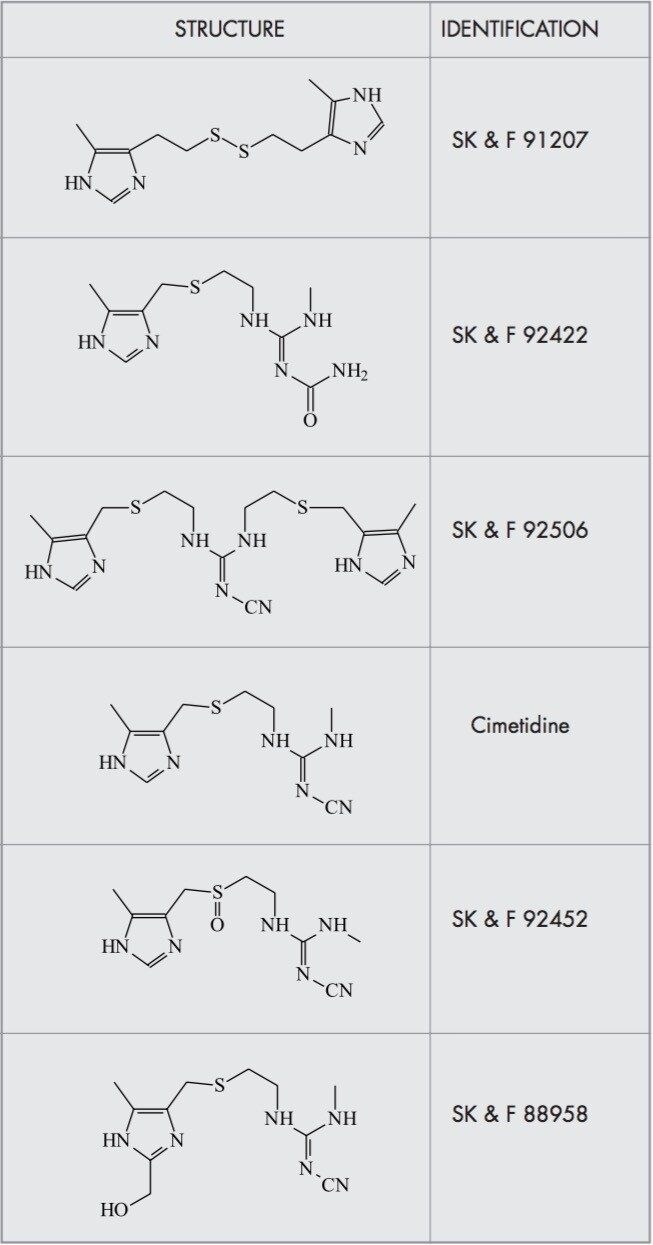
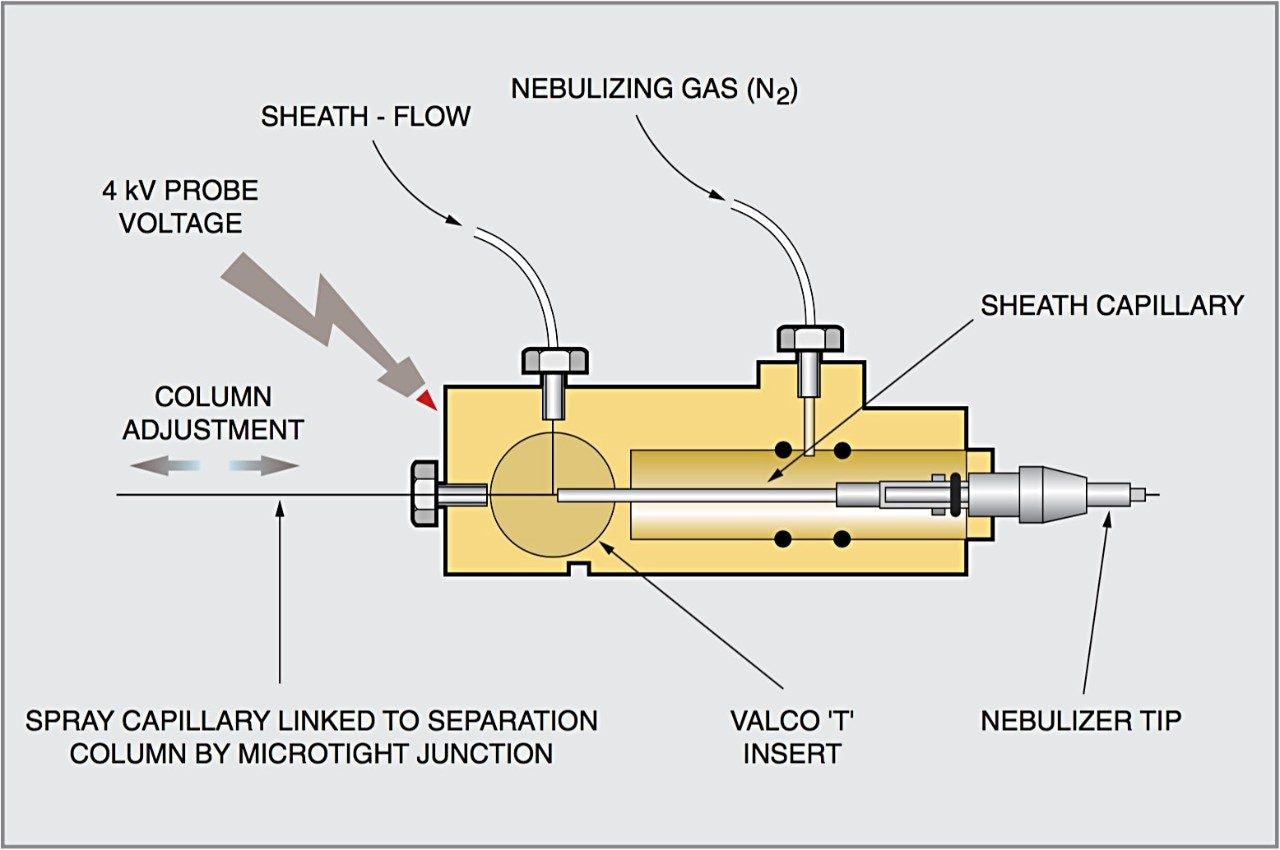
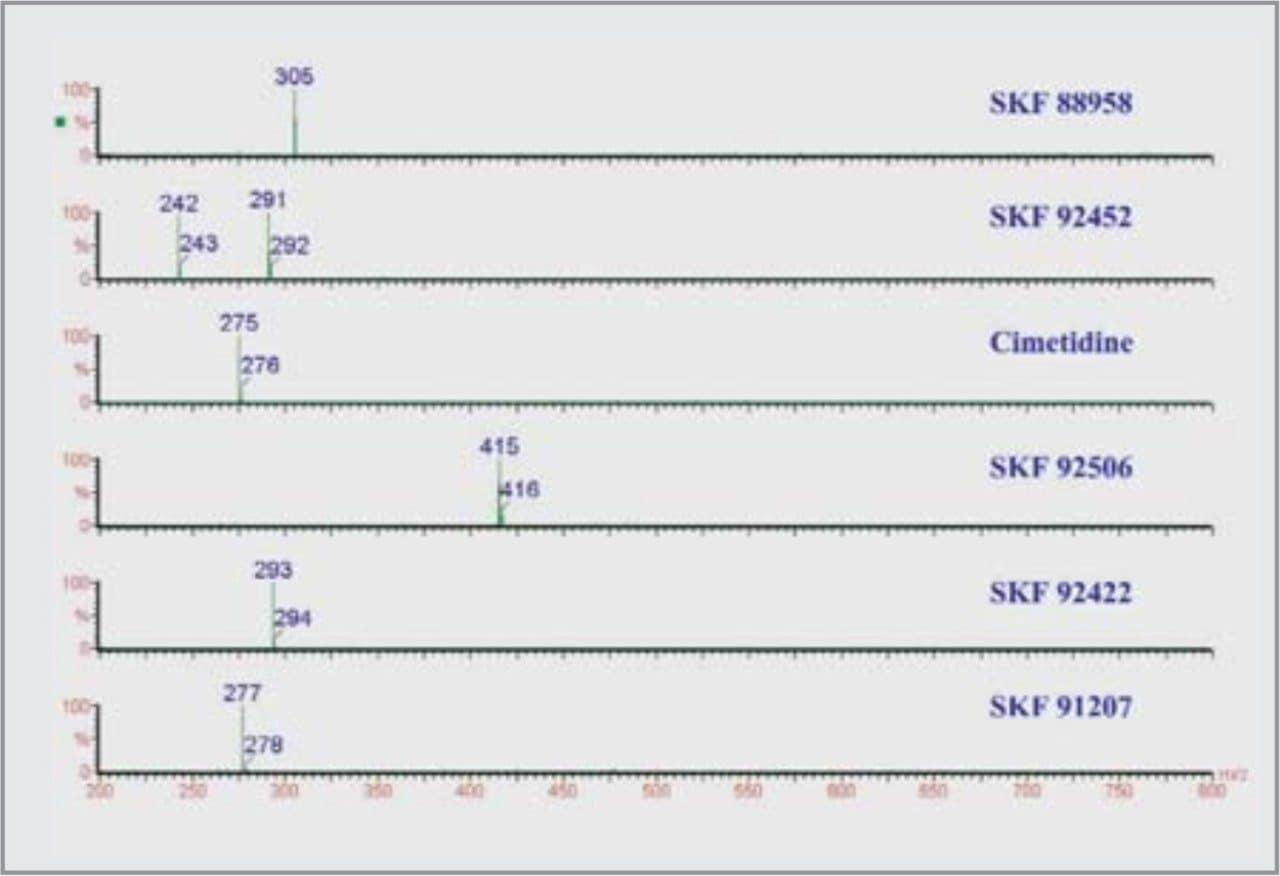
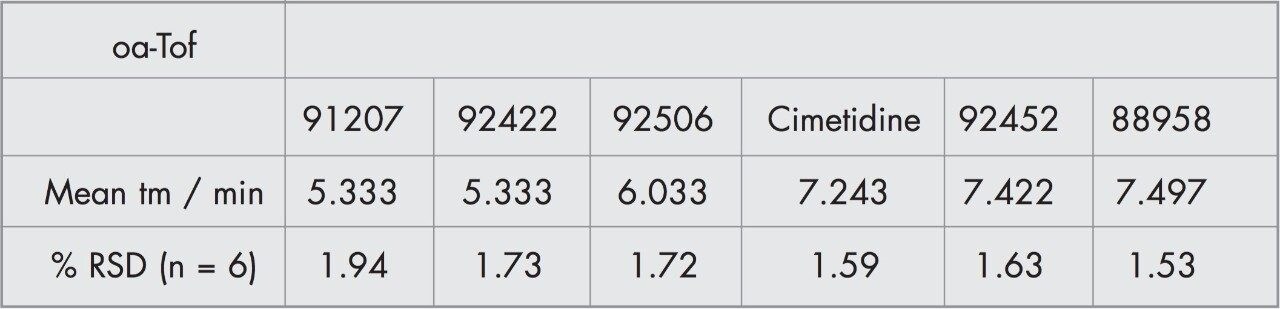
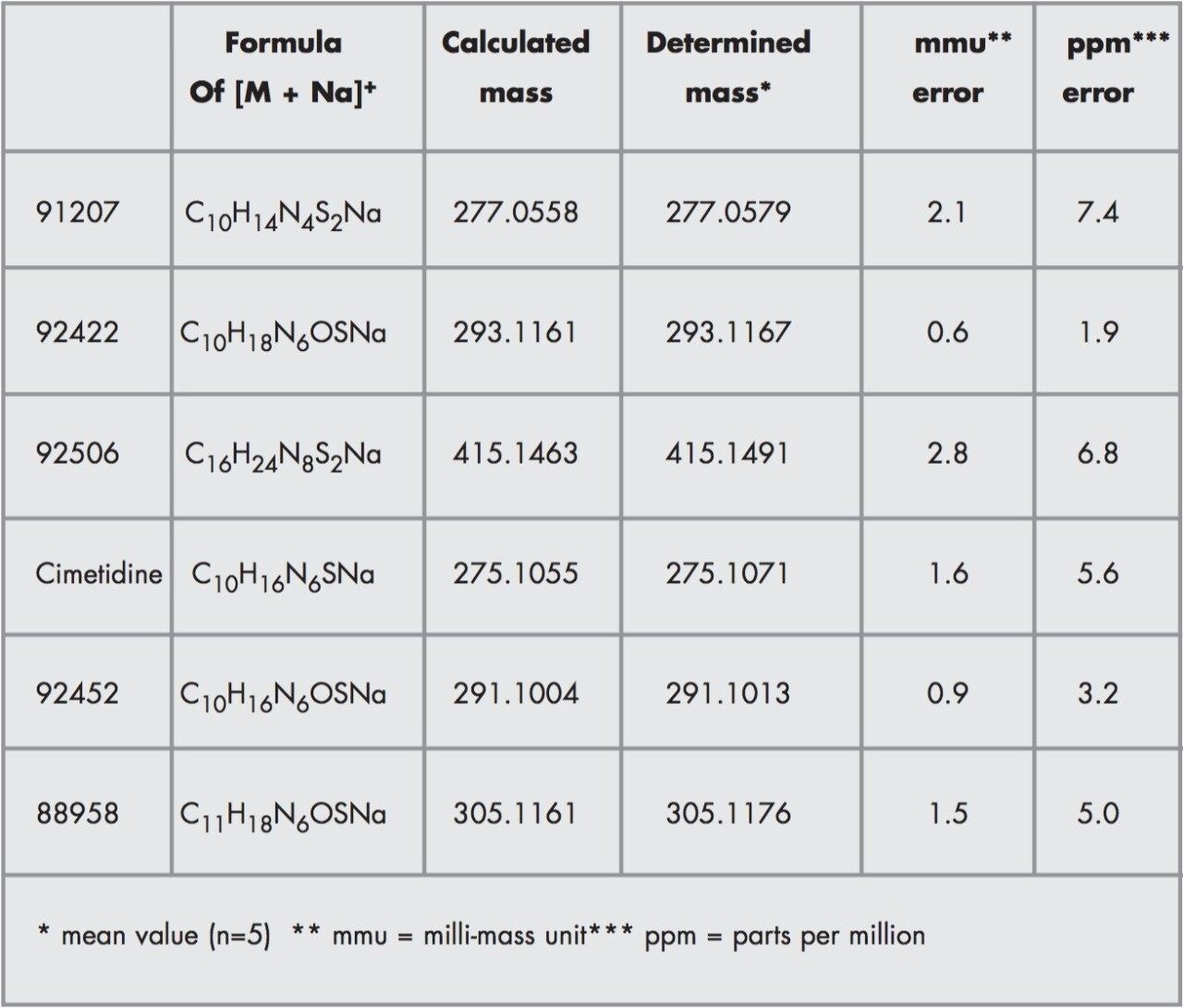
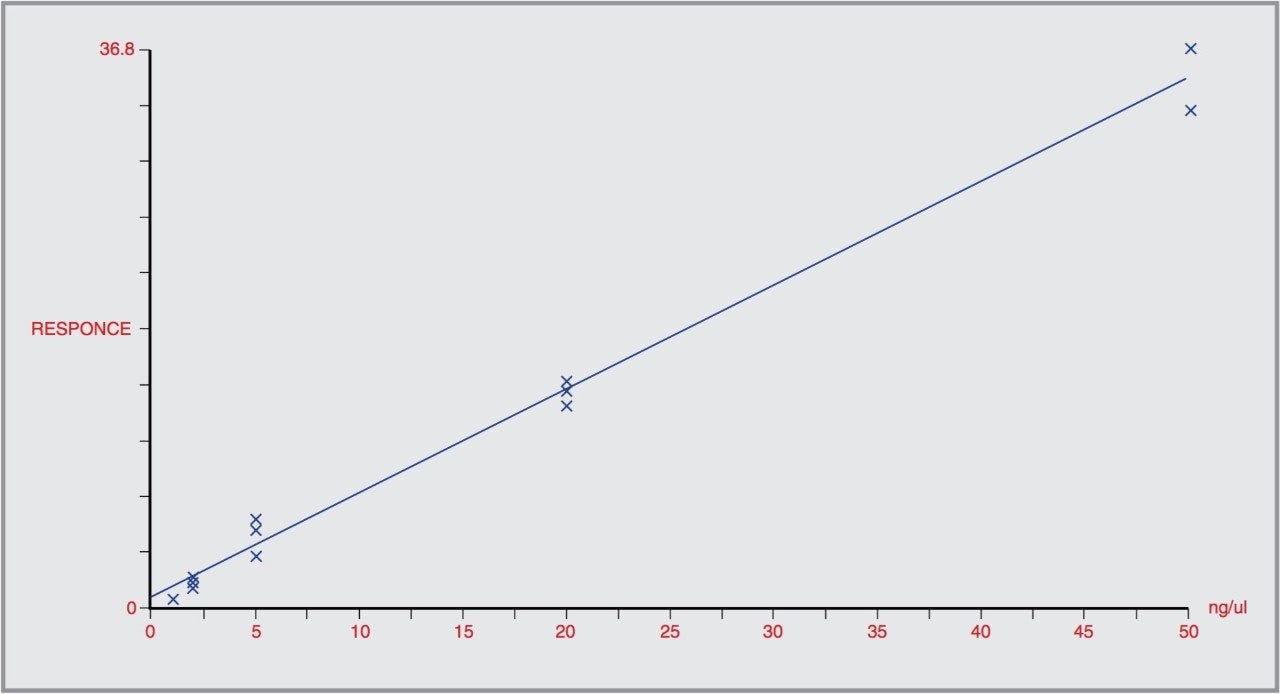
CZE-ToF MS has recently been successfully applied to the analysis of standard peptide and protein mixtures1-6. Accurate mass measurement of electrophoretic peaks has also been achieved to aid protein identification by peptide mapping1. However to the best of the author’s knowledge this is the first example of accurate mass measurement of low molecular weight pharmaceutical compounds using CZE-oa-ToF mass spectrometry.
Separations in CZE are obtained by differential migration of charged solutes through a capillary column when under the influence of an electric field. This separation occurs because of differences in the electrophoretic mobilities of analyte ions. Electrophoretic mobility is based on charge-to-size ratio (i.e. a highly charged, small ion will migrate faster than a lesser charged, large ion). A feature of CZE is the electroosmotic flow (EOF), which is the bulk flow of liquid through the column. The EOF arises through ionic interaction with the capillary wall, these ions move (and drag the bulk liquid) through the column by electrostatic attraction when the separation voltage is applied. The EOF through the capillary has a flat-flow profile (unlike the laminar flow associated with pressure flow techniques), which results in lower solute zone diffusion and extremely efficient separations, (plate numbers/metre in excess of 1 x105 are common).
Peak widths produced are of the order of a few seconds, requiring very fast acquisition rates and high sensitivities (usually ng quantities are injected onto the capillary). Scanning instruments may have difficulty in obtaining sufficient data points across such narrow peaks (although the use of selected ion monitoring (SIM) mode may overcome this). ToF MS has distinct advantages over scanning instruments, these include sensitivity (due to high duty factors, leading to a greater percentage of the ions formed being detected), fast acquisition rates (upto 10 spectra s-1), and high m/z range. oa-ToF mass spectrometers seem ideally suited to being coupled with capillary zone electrophoresis (CZE).
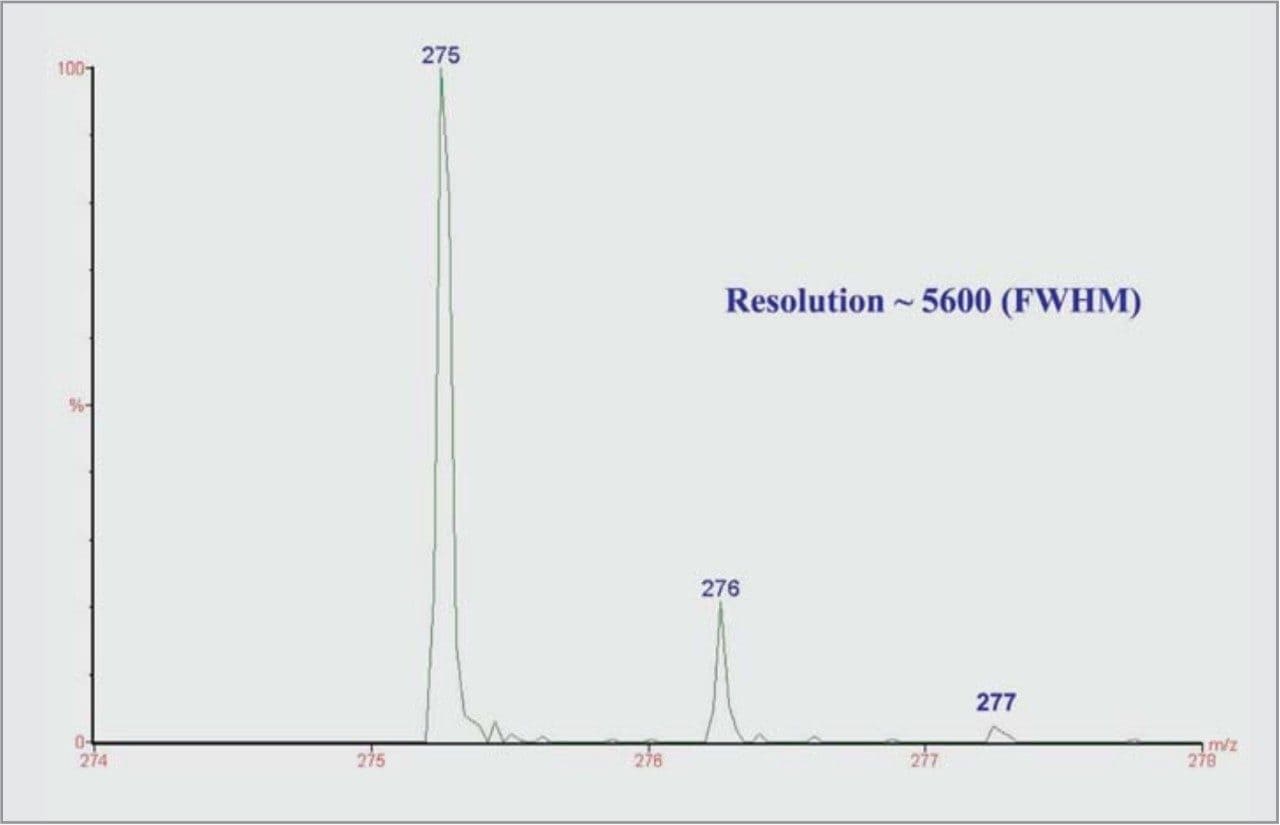
The Micromass LCT is an oa-Tof instrument which is capable of resolutions of 5000 full width half maximum (FWHM) and gives mass measurement accuracies of better than 5ppm, allowing the molecular formula to be predicted or confirmed.
We have investigated the use of the Micromass LCT coupled to CZE. The on-line separation of a drug substance and related impurities has been achieved.