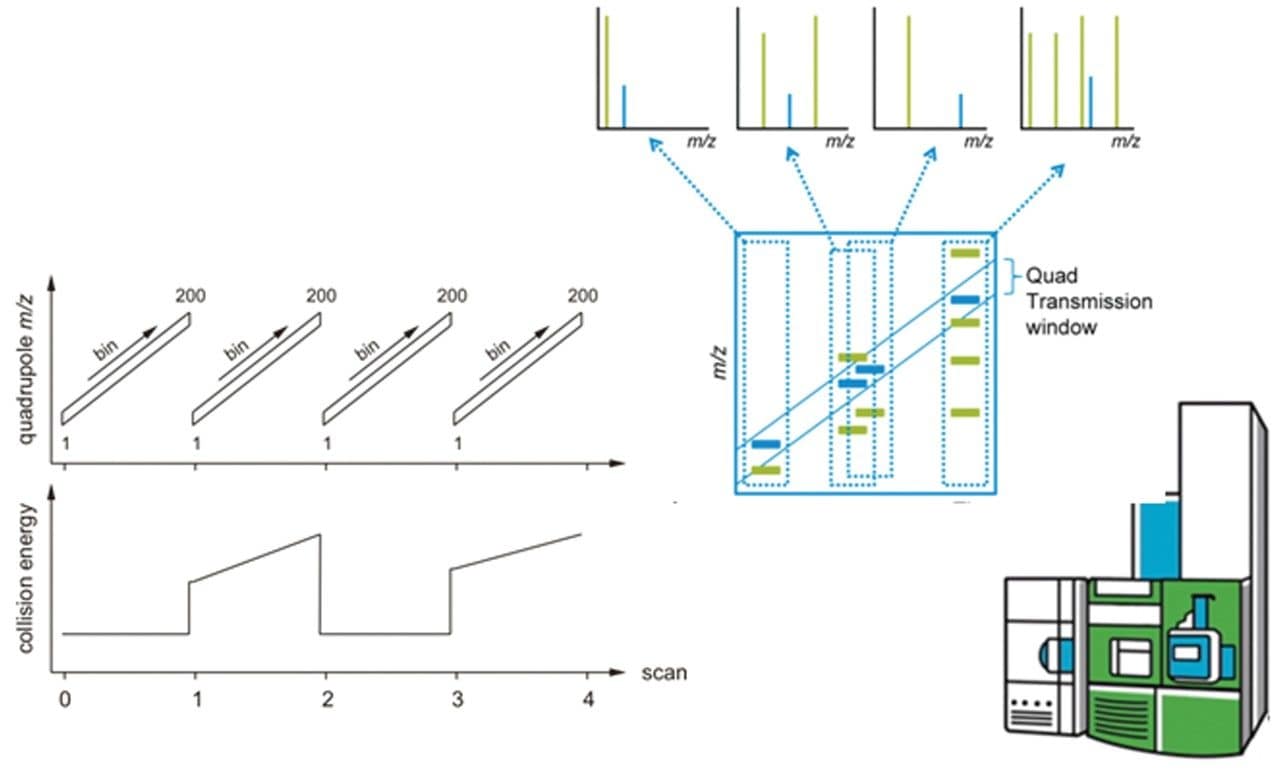
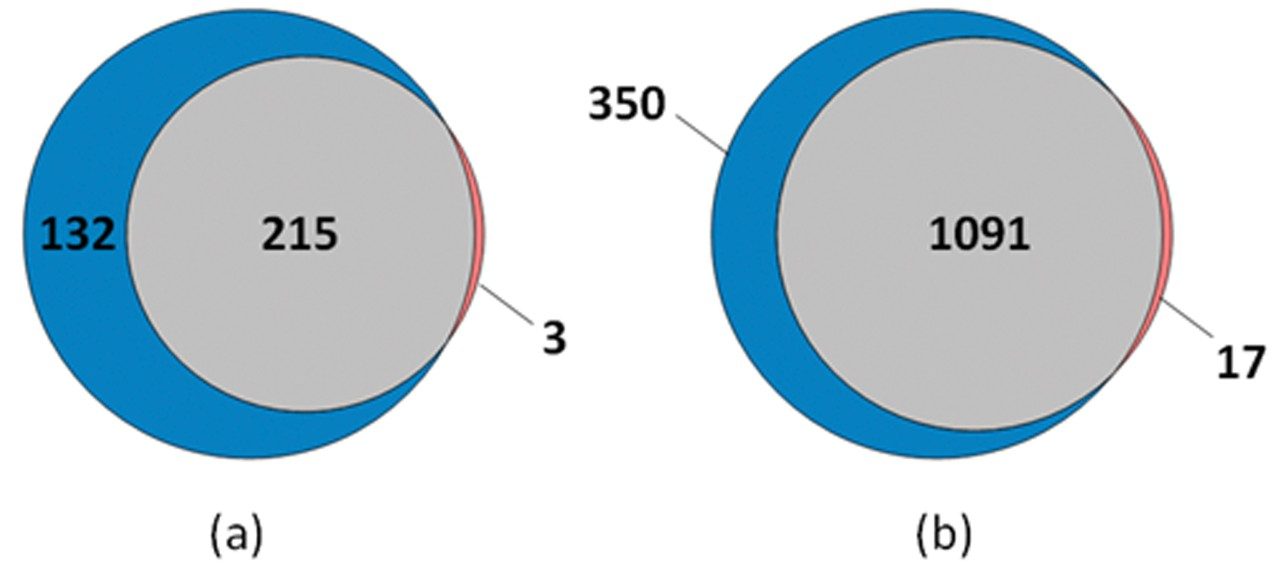
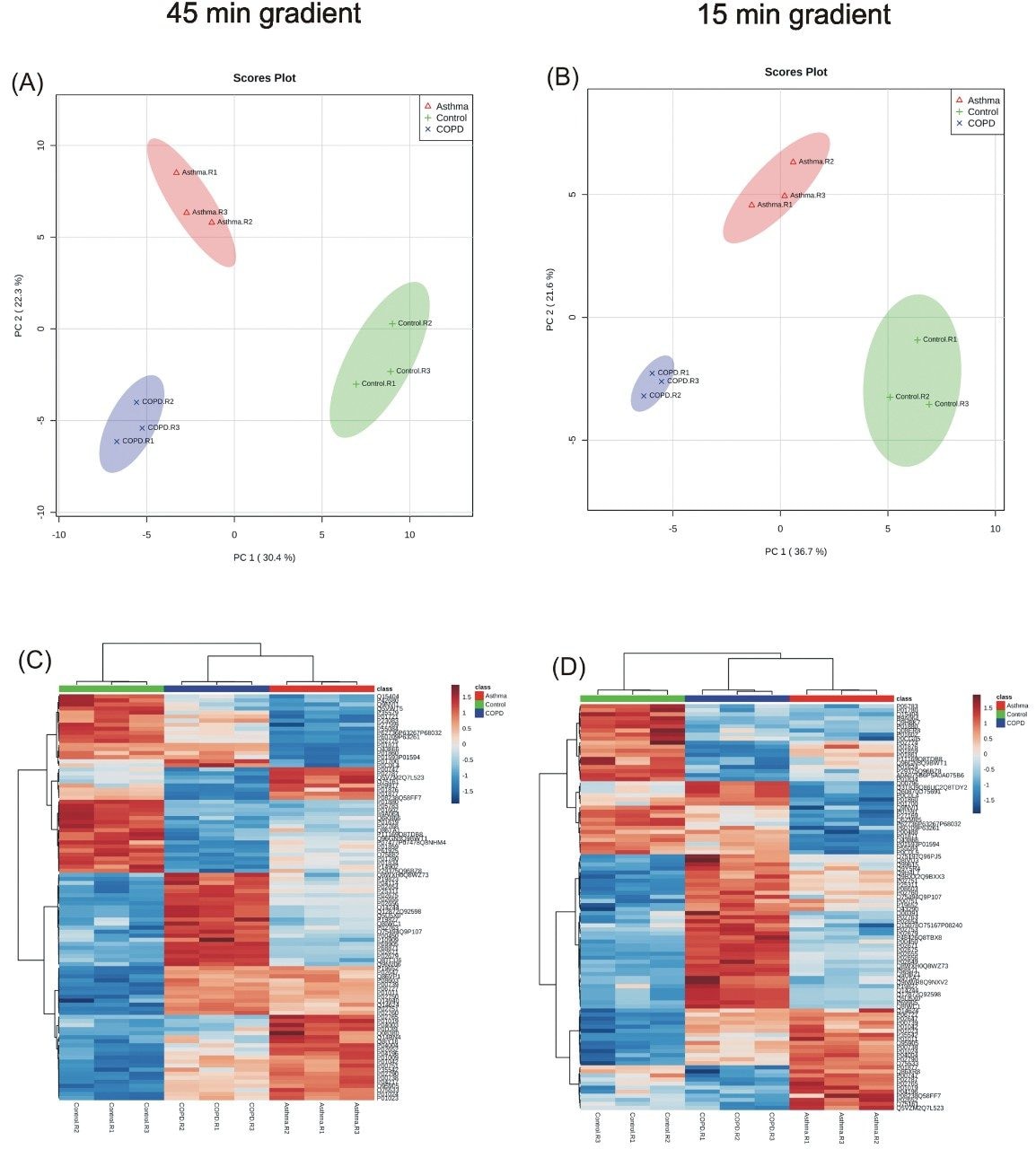
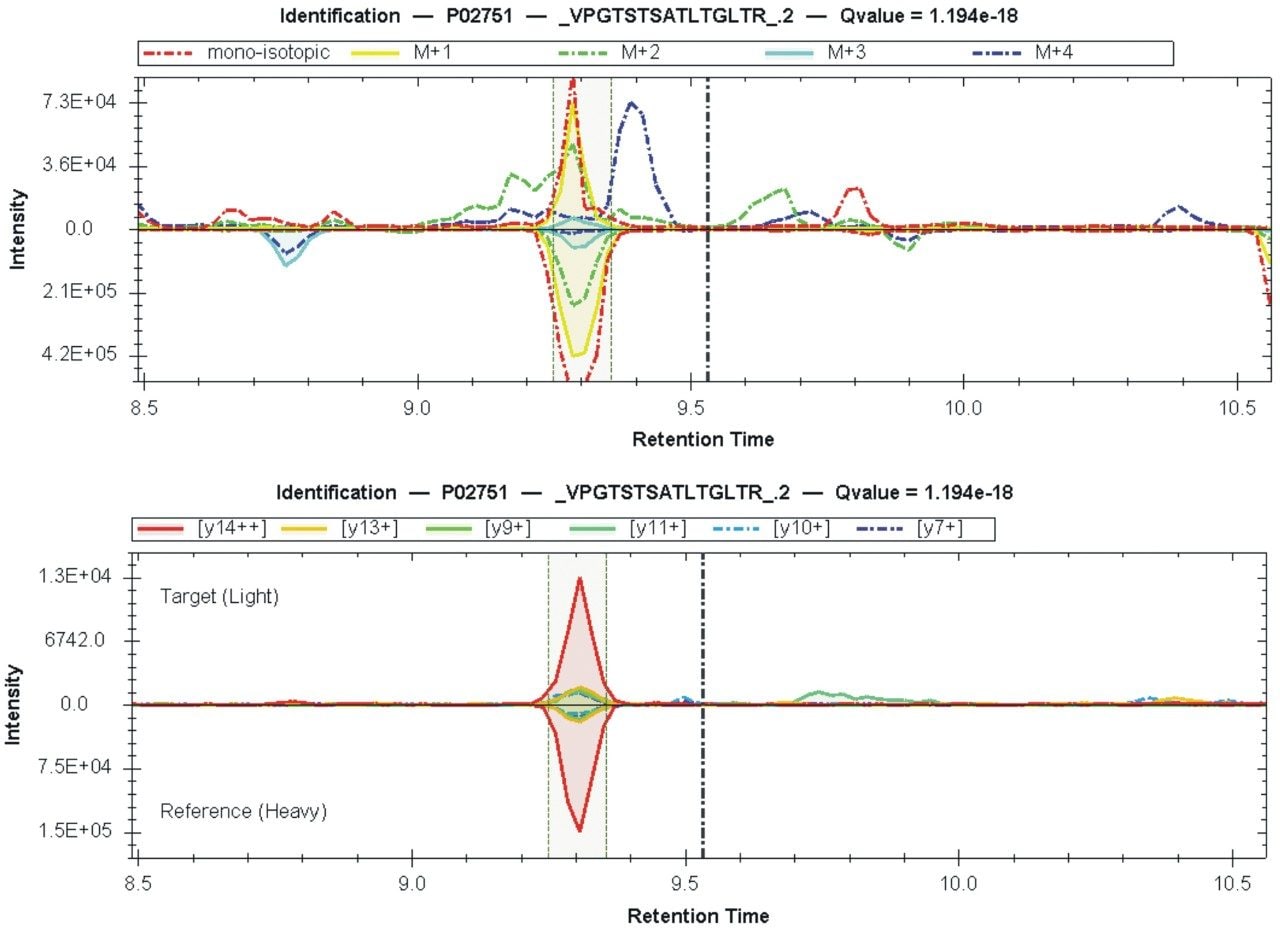
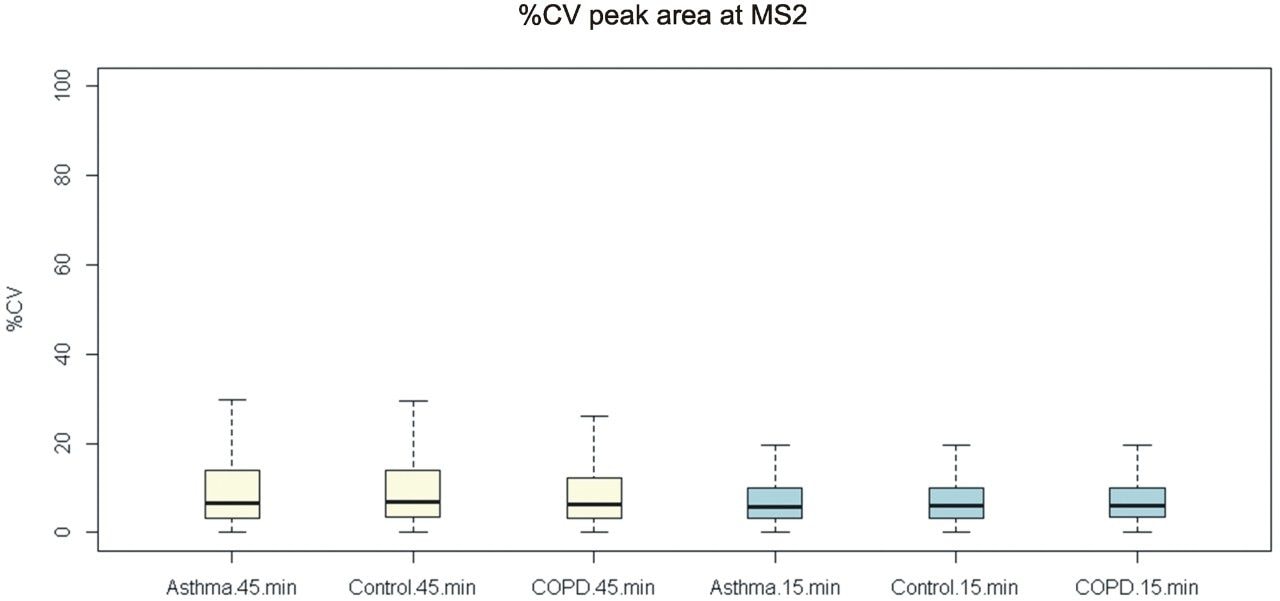
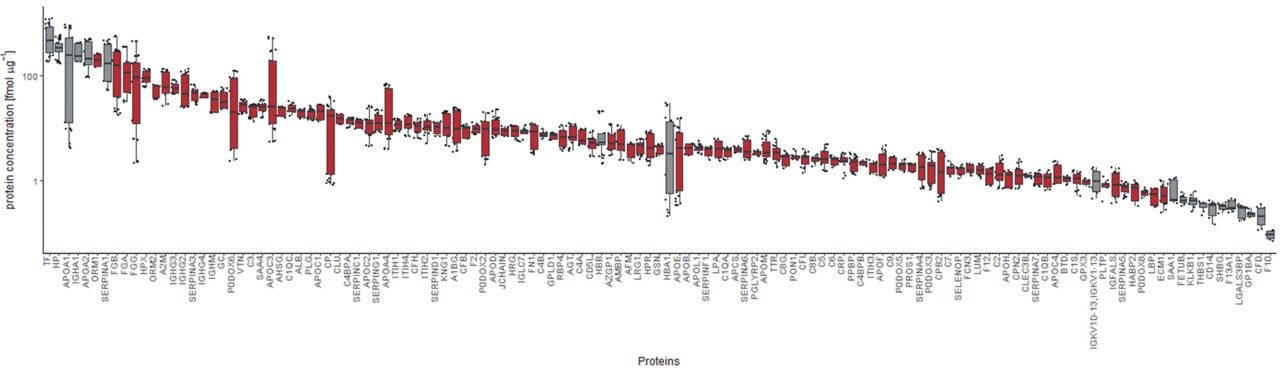
Quantitative proteomics often incorporates the use of stable labeled isotopes (SILs) in order to provide absolute quantification. Recent advancements have seen the introduction of peptide panels allowing the quantification of more than 500 proteins in plasma sample sets. However, this is technically challenging when attempting to acquire the data using more traditional MS acquisition modes such as multiple reaction monitoring (MRM), since the duty cycle of the instrument is compromised and therefore results in under sampling of the data. An alternative approach is to apply SONAR, a data independent analysis (DIA) methodology, allowing for high throughput while also ensuring high specificity and maintaining quantitative performance. SONAR has previously been described,1,2 highlighting the utilization of a fast scanning quadrupole, enabling the technique to be compatible with fast chromatography, high throughput workflows. Clinical research using proteomics is one high throughput example which requires the analytical benefits provided by SONAR. Here, we present the applicability of SONAR for high throughput, absolute quantification of plasma proteins from human samples consisting of controls, chronic obstructive pulmonary disease (COPD), and asthma.