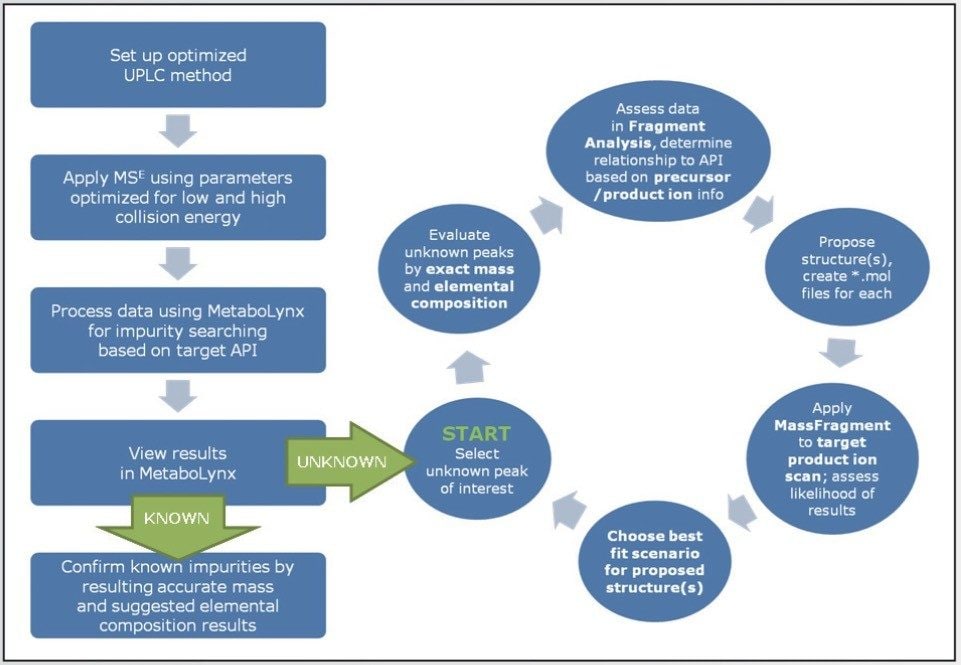
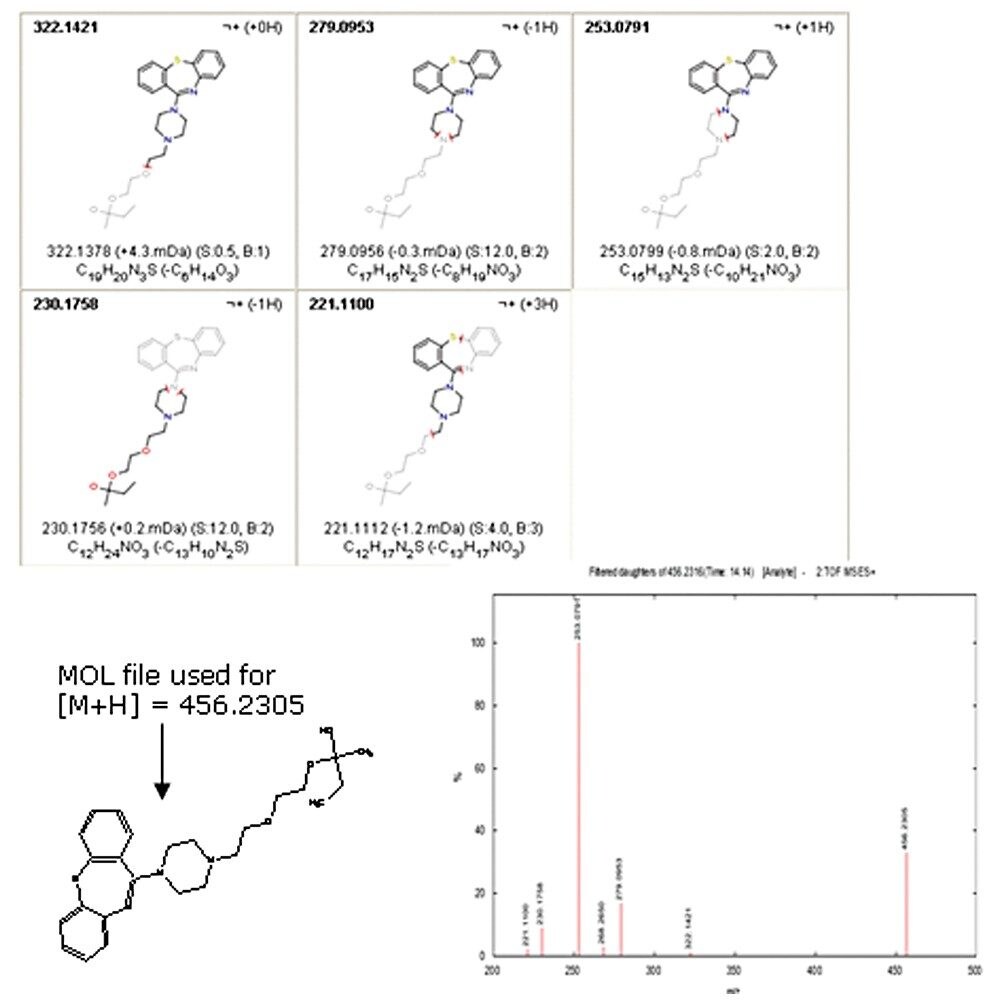
Evaluating known and unknown impurities
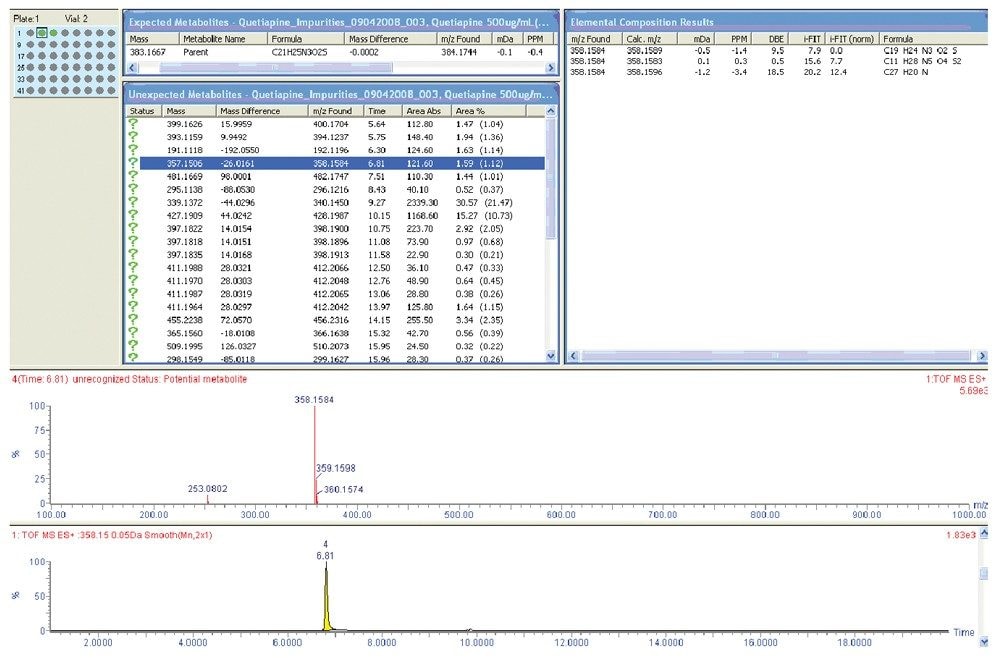
Evaluation of the unknown impurity peaks by exact mass and elemental composition of quetiapine hemifumarate using MetaboLynx XS indicated that the mass accuracy of the API quetiapine was reported to be 0.4 ppm. A total of 80 impurity peaks were listed. Upon adjustments to integration and data filtering, 44 peaks were found to be relevant. Non-relevant peaks were observed to be anomalies of initial integration of noise and peaks with extremely low-level response in UV and MS detection.
Ten known impurities were observed with an average mass accuracy of 1.3 ppm. Two known masses, 398.19xx and 412.20xx, had three and four separate retention times listed, respectively. The masses with multiple chromatographic retention times, which indicated possible structural isomers, were:
[M+H] = 398.19xx observed four peaks, three of which met the reporting threshold. The observed [M+H] = 398.1900, 398.1896, 398.1913 at retention times (RT) of 10.75 min., 11.08 min., and 11.58 min., with measured mass accuracies of 0.5 ppm, 1.5 ppm, and 2.8 ppm, respectively, resulted in an identified elemental composition of C22H28N3O2S
[M+H] = 412.20xx observed five peaks, four of which met the reporting threshold. The observed [M+H] = 412.2066, 412.2048, 412.2065, and 412.2059 at retention times (RT) of 12.50 min, 12.76 min, 13.06 min, and 13.97 min, with measured mass accuracies of 1.7 ppm, 2.7 ppm, 1.5 ppm, and 4.1 ppm, respectively, resulted in an identified elemental composition of C22H29N3O2S. In terms of the unknowns that were identified, of 21 entries for 15 chromatographic peaks:
Peaks identified as doubly charged species:
- [M+2H]2+ = 353.1512, [M+H]+ 705.3013 at RT = 17.20 min
- [M+2H]2+ = 309.1256, [M+H]+ 617.2514 at RT = 17.36 min
- [M+2H]2+ = 684.2089 with a large fragment at [M+H] = 382.3485
Peaks with multiple m/z ions; which could be possible coelutions, included:
- Peak RT = 15.96 min observed [M+H] = 510.2073, 299.1627, 399.2523 (three intense m/z values)
- Peak RT = 17.42 min observed [M+H] = 653.3301, 592.1955 (two intense m/z values)
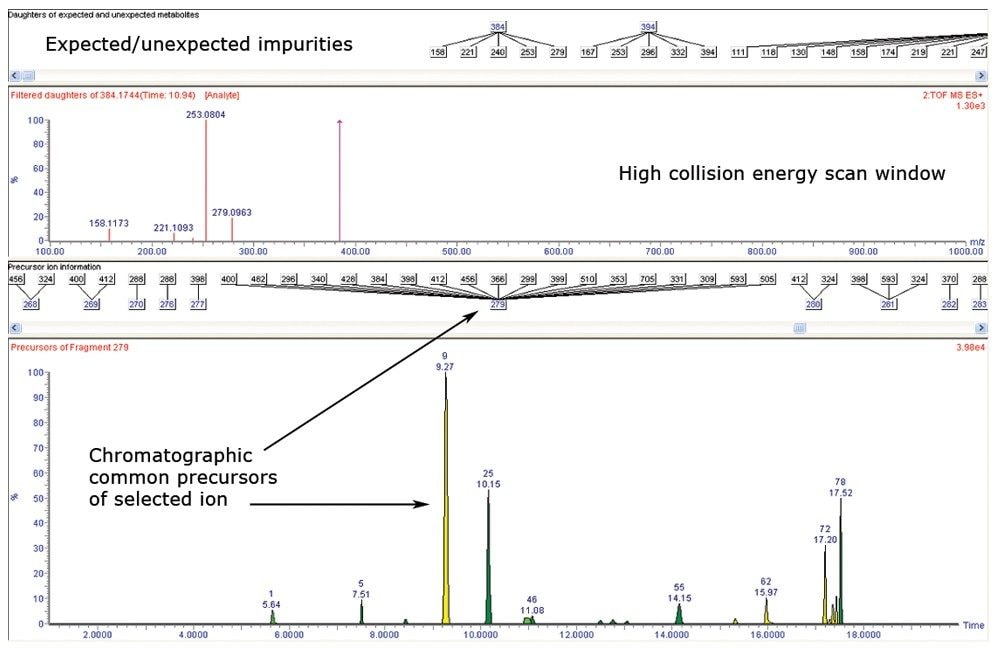
From these data, we can generate and assess the data in the Fragment Analysis function of MetaboLynx XS by determining the relationship to the API based on the MSE precursor/product ion information.