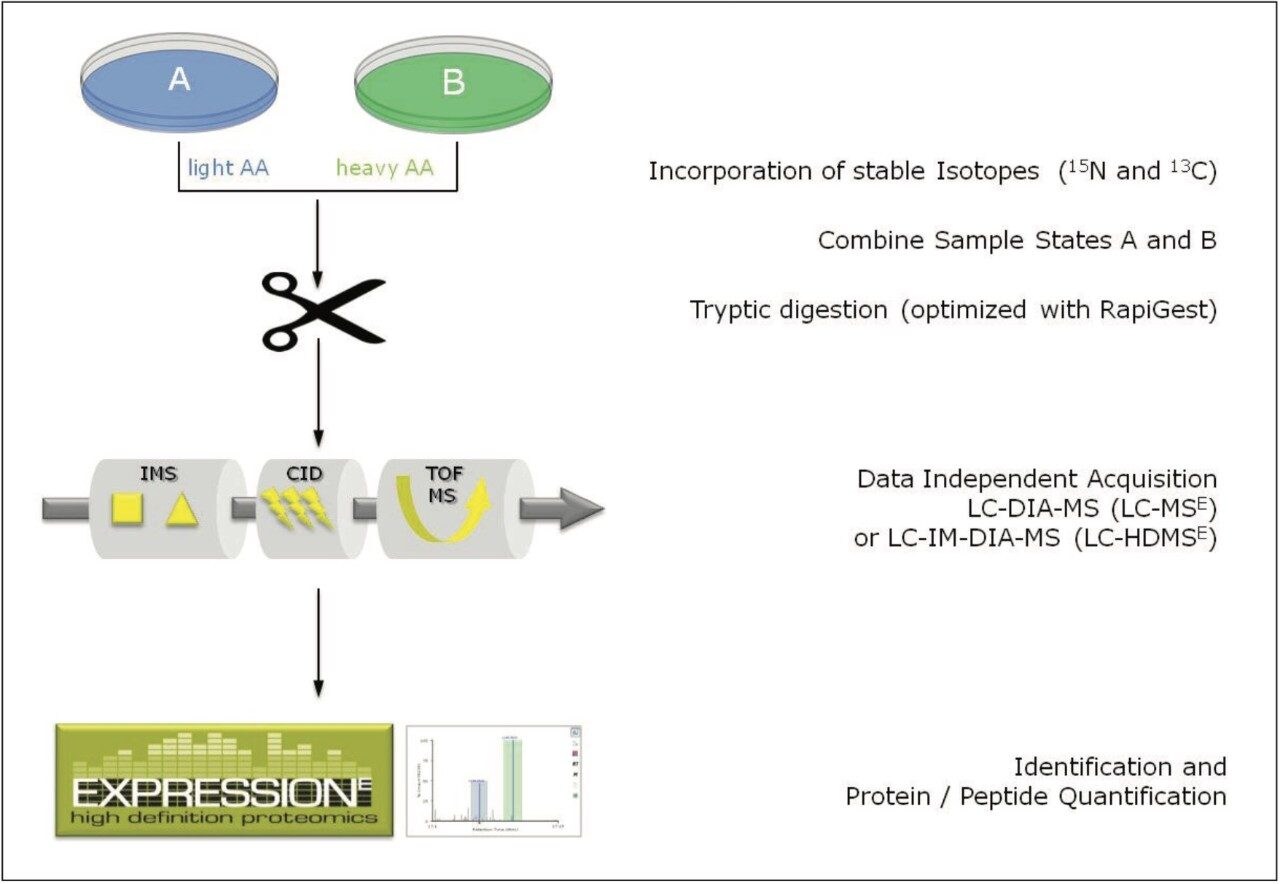
Stable isotope labeling with amino acids in cell culture (SILAC) is a popular metabolic labeling technique for mass spectrometric (MS)-based quantitative proteomics.1 In SILAC, cell cultures to be compared are grown in culture medium containing either naturally occurring, or stable-isotope labeled amino acids. Over several divisions, the amino acids are incorporated into cellular proteins, enabling the comparison of amounts of particular proteins from one cell type to its comparator.
In a comparative shotgun proteomics experiment, differentially labeled samples are mixed early in the experimental process and analyzed together by LC-MS/MS. The labeling does not affect the chemical properties of peptide; hence, they coelute from the LC column and can be analyzed simultaneously in the mass spectrometer. The peptides of the differentially labeled samples can be accurately quantified relative to each other to determine relative protein abundances.
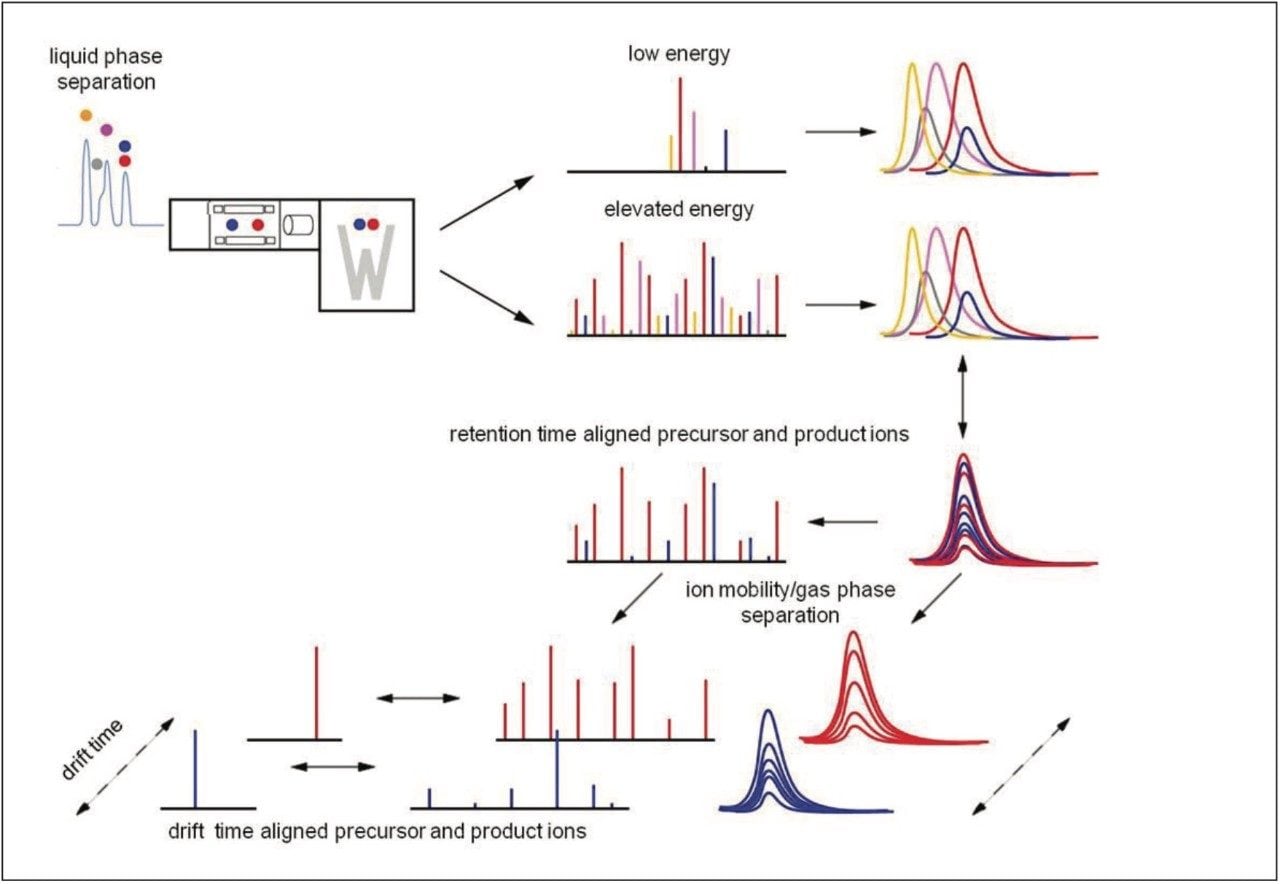
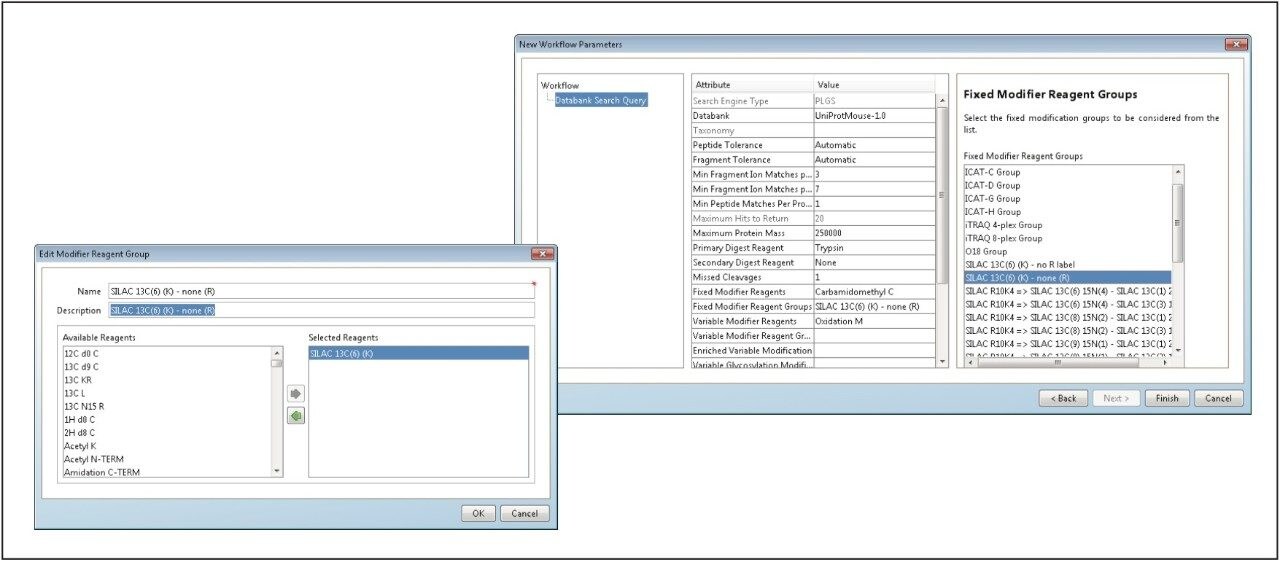
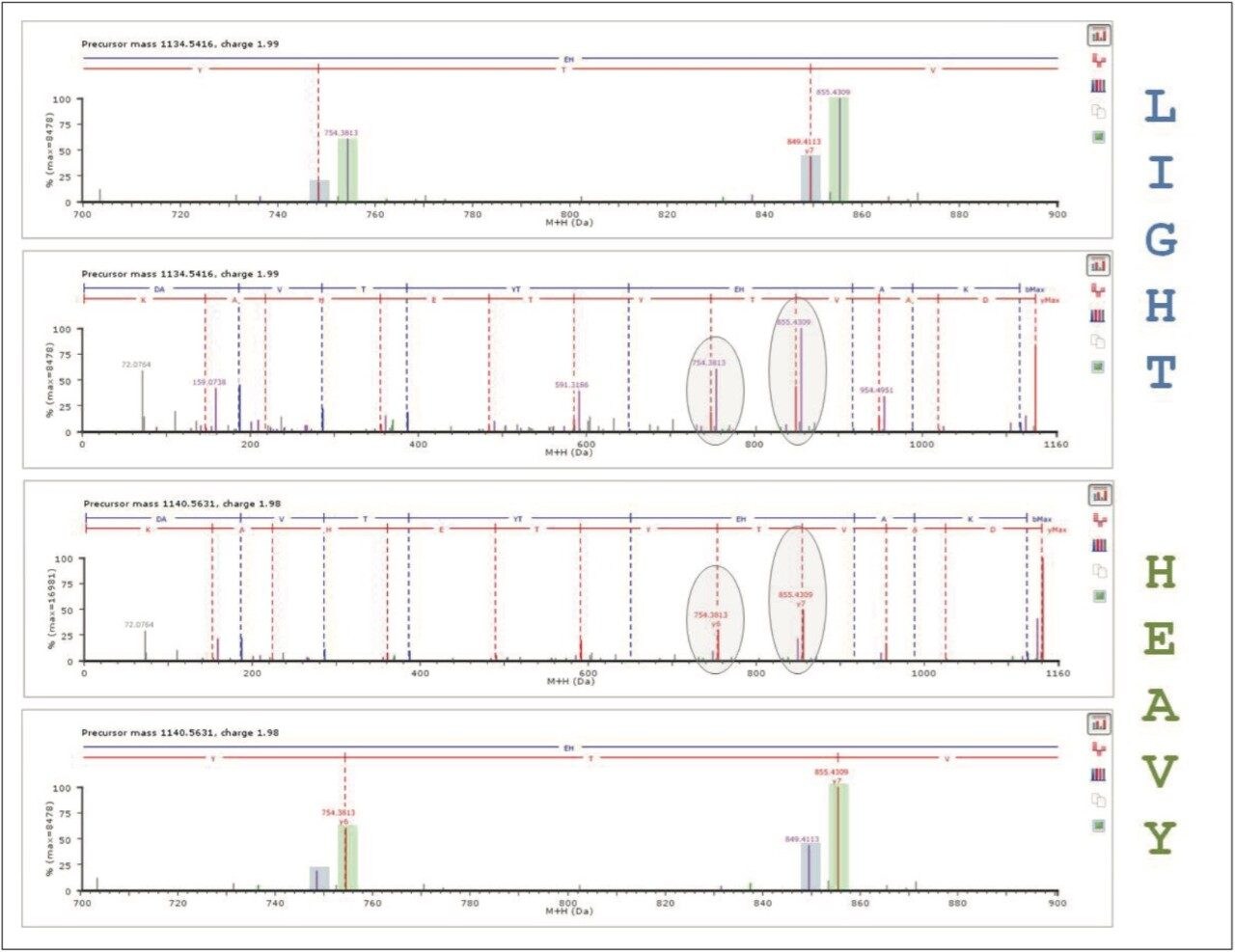
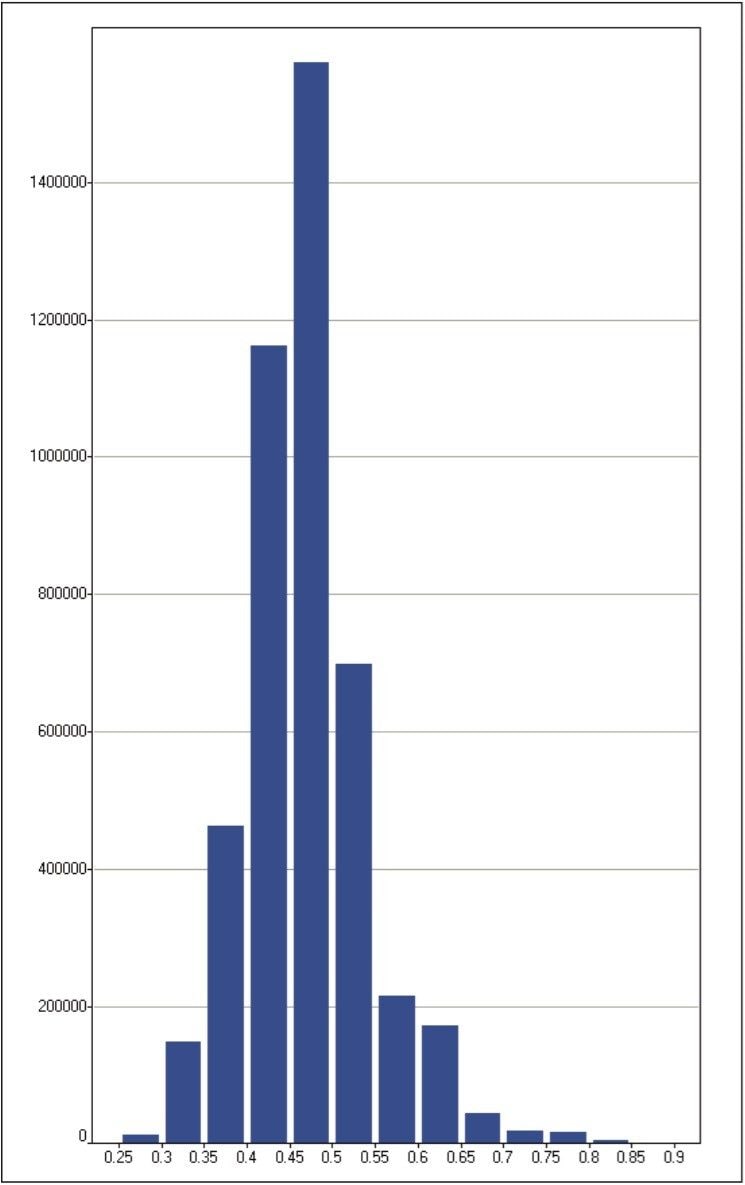
To date, the SILAC method has been predominantly applied to analyze cell cultures using data-dependent analysis (DDA) MS/MS techniques. However, DDA approaches are limited by issues with chimericy, a phenomenon likely to be more problematic in the increasingly complex samples observed in SILAC experiments.2 In this investigation, ion mobility (IM)-enabled data-independent analysis (DIA) has been utilized to quantify SILAC-labeled mutant embryonic mouse cells.
This application note demonstrates the functionality of hardware and software, along with the accuracy and precision obtained for the quantitative analyses of SILAC-labeled samples using LC/IM-DIA-MS (HDMSE) on a SYNAPT G2-S Mass Spectrometer