Understanding the occurrence, fate, and impact of persistent organic pollutants (POPs) is a global priority and consequently is undertaken by a diverse range of organizations. Perfluorinated compounds (PFCs) have become increasingly important and perfluorooctane sulphonic acid (PFOS) has been included in the Stockholm Convention on POPs.1 The tracking of PFCs is critical to organizations whose activities might inadvertently facilitate exposure to populations through water, food supplies, and consumer products. This is also a priority for researchers and regulators and is especially true when taking into account the high publicity related to newer contaminants such as PFCs.
Over the past decade these compounds have been determined in an array of matrices by various techniques with liquid chromatography tandem quadrupole mass spectrometers (LC-MS/MS) featuring heavily.2 The ability of laboratories to successfully measure PFCs in various matrices has improved greatly in recent times, largely due to improvements in labeled standard availability highlighted in recent inter laboratory studies.3,4 These studies also attribute the continuous improvement in data quality to advances in instrumental technology. Advances in LC-MS/MS instrument performance have largely been focused on Multiple Reaction Monitoring (MRM) sensitivity to satisfy the need for increasingly lower detection limits. While this is clearly a priority for this type of instrumentation, there have been limitations previously in acquiring important qualitative information from a sample in a single injection with previous generation instruments.
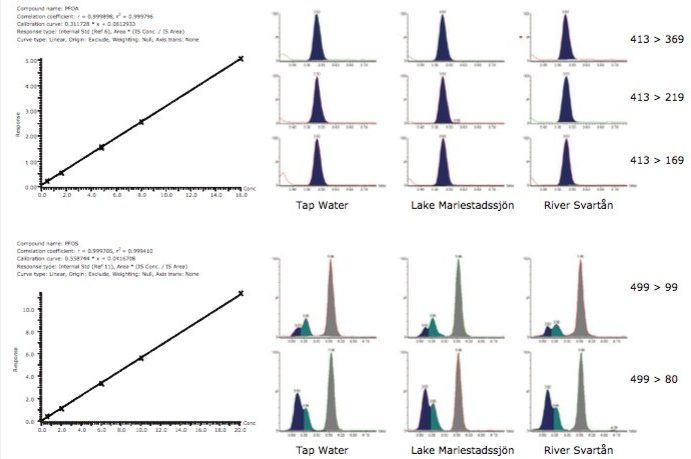
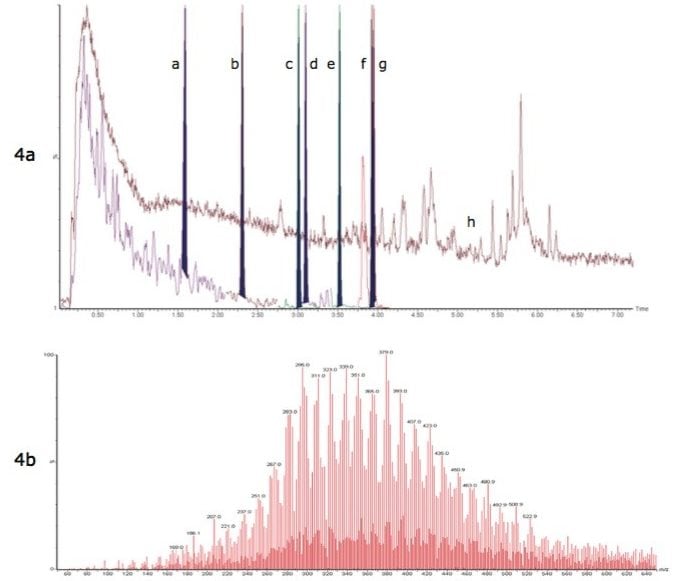
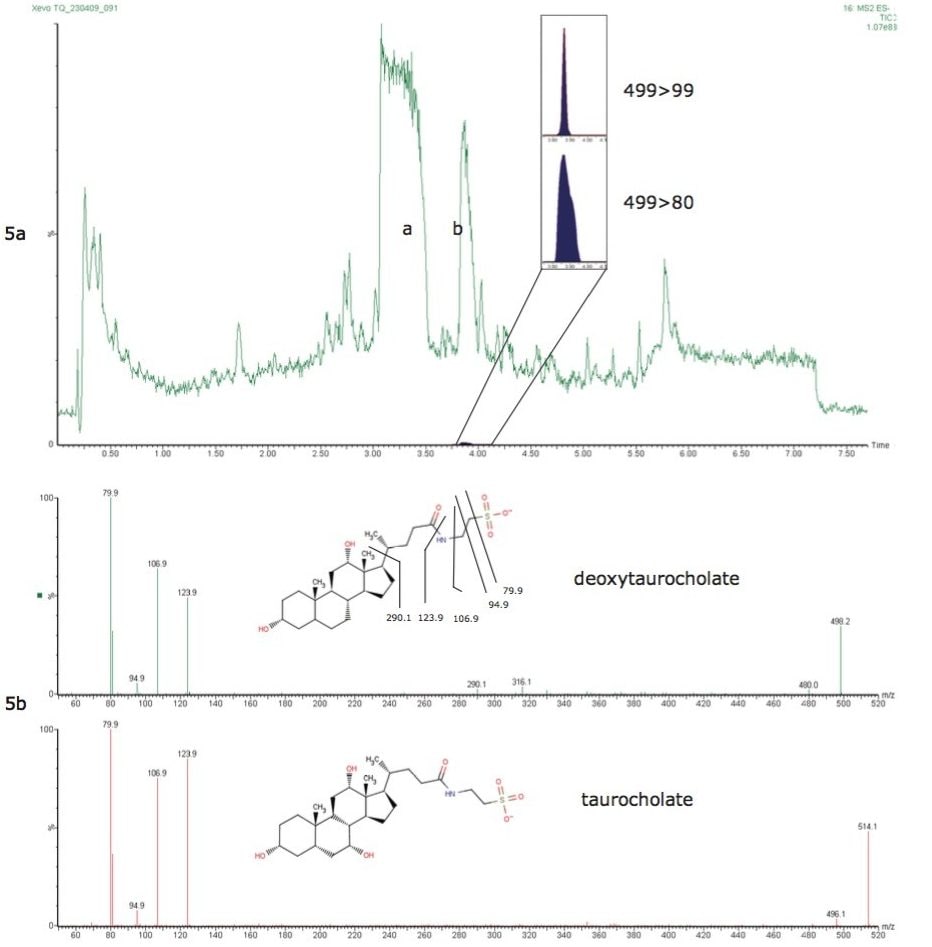
This information can be of high value as a method development tool or intra-sample QC check when analyzing ultra-trace level contaminants in difficult sample matrices such as environmental waters and biological tissues. This ultimately leads to greater confidence in the analytical result.
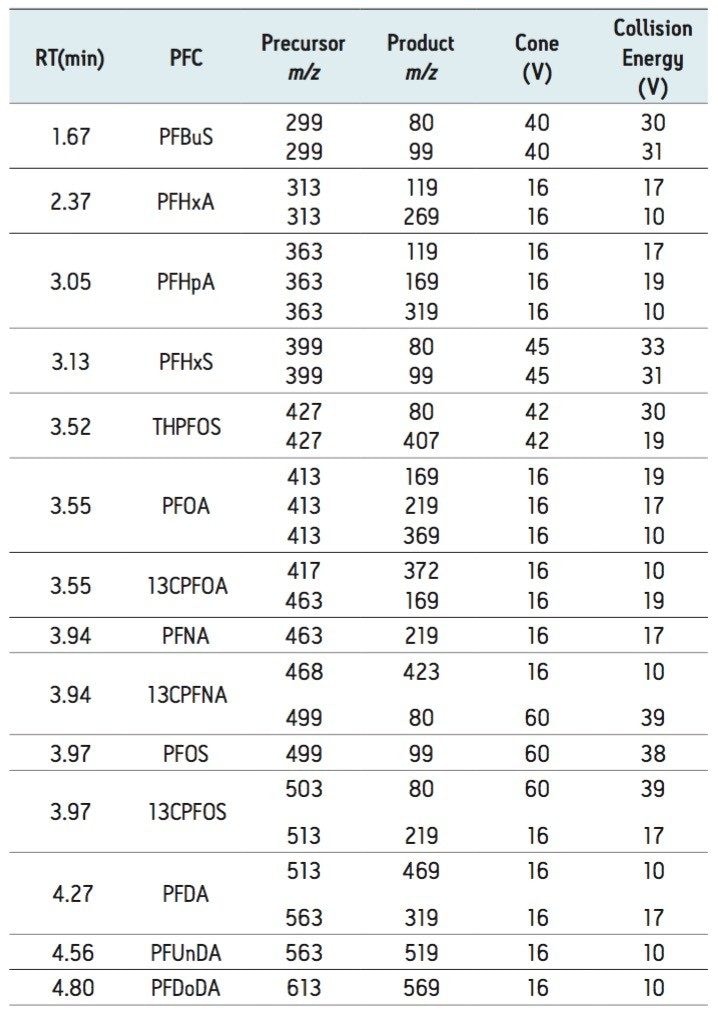
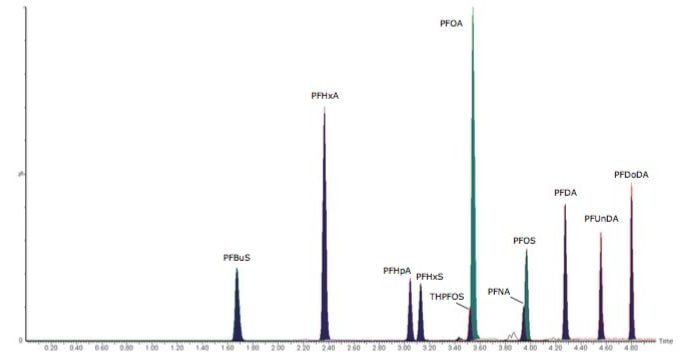
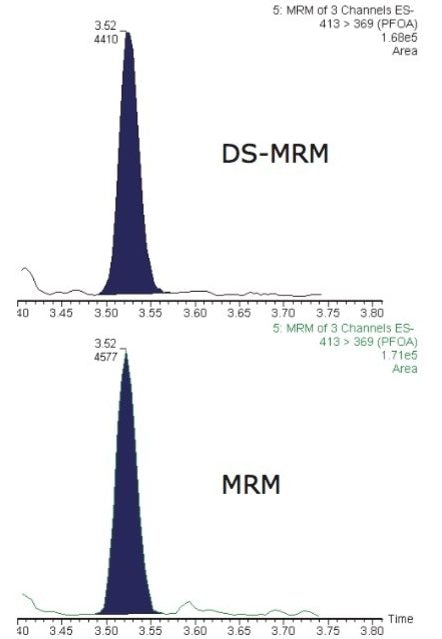
This application note describes advanced high-sensitivity Xevo TQ-MS analysis of PFCs in environmental waters and biota, incorporating Waters RADAR dual scan-MRM functionality to understand the nature of the sample matrix, while simultaneously performing routine quantitation.