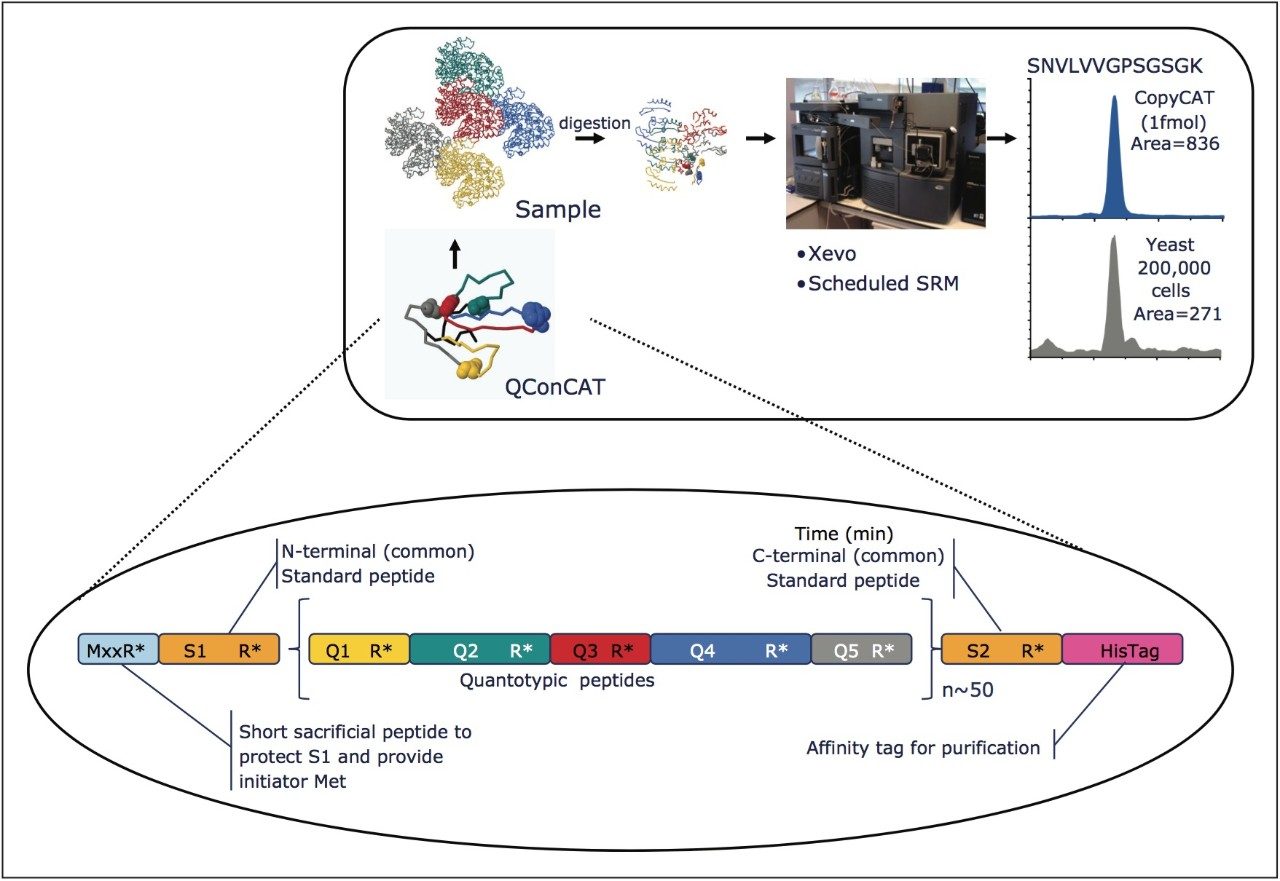
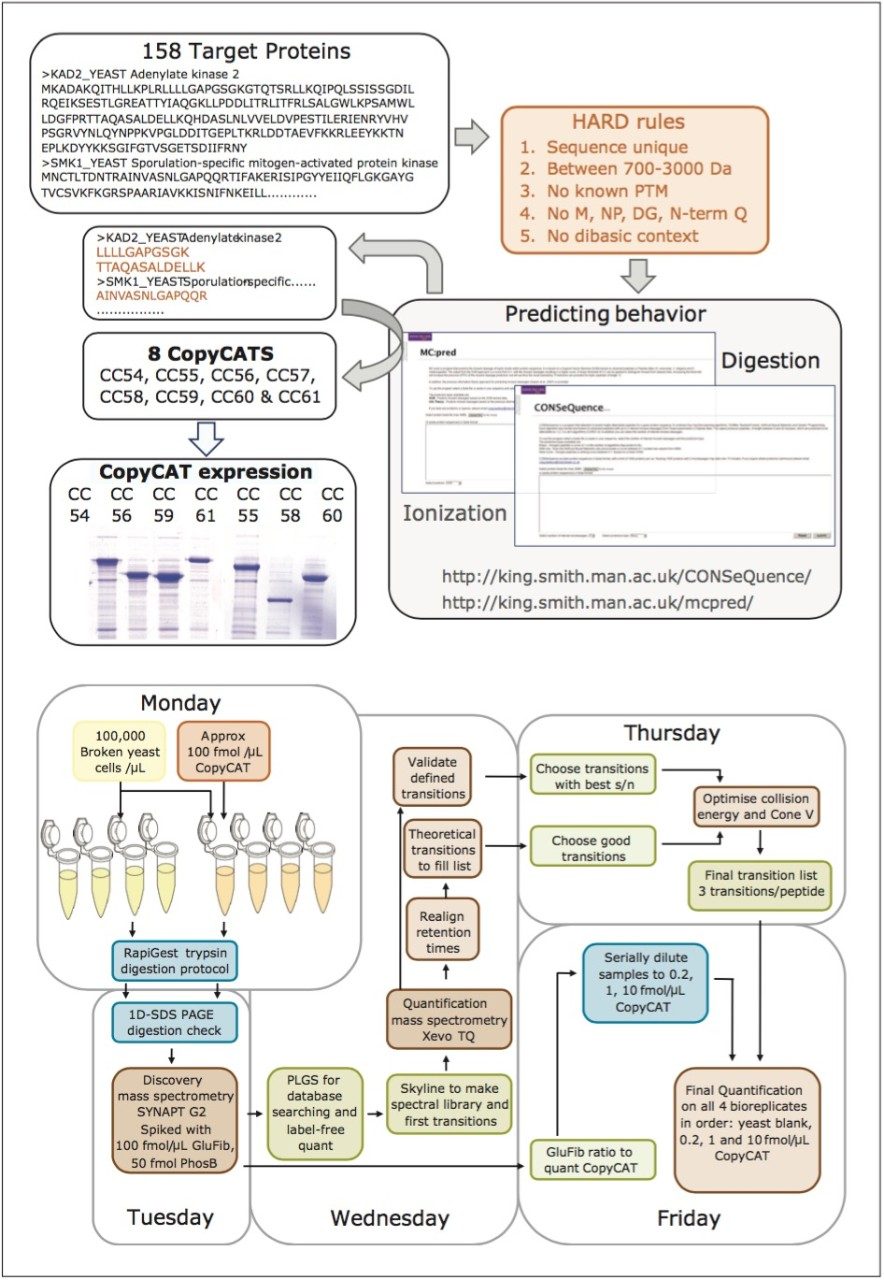
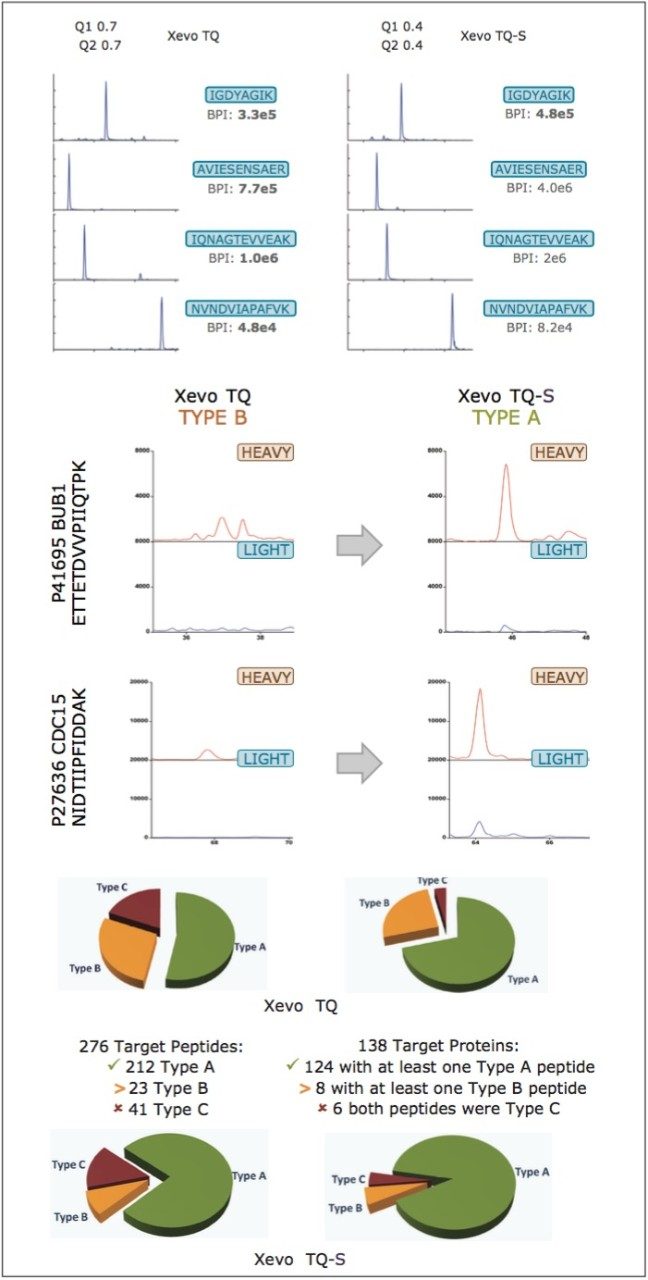
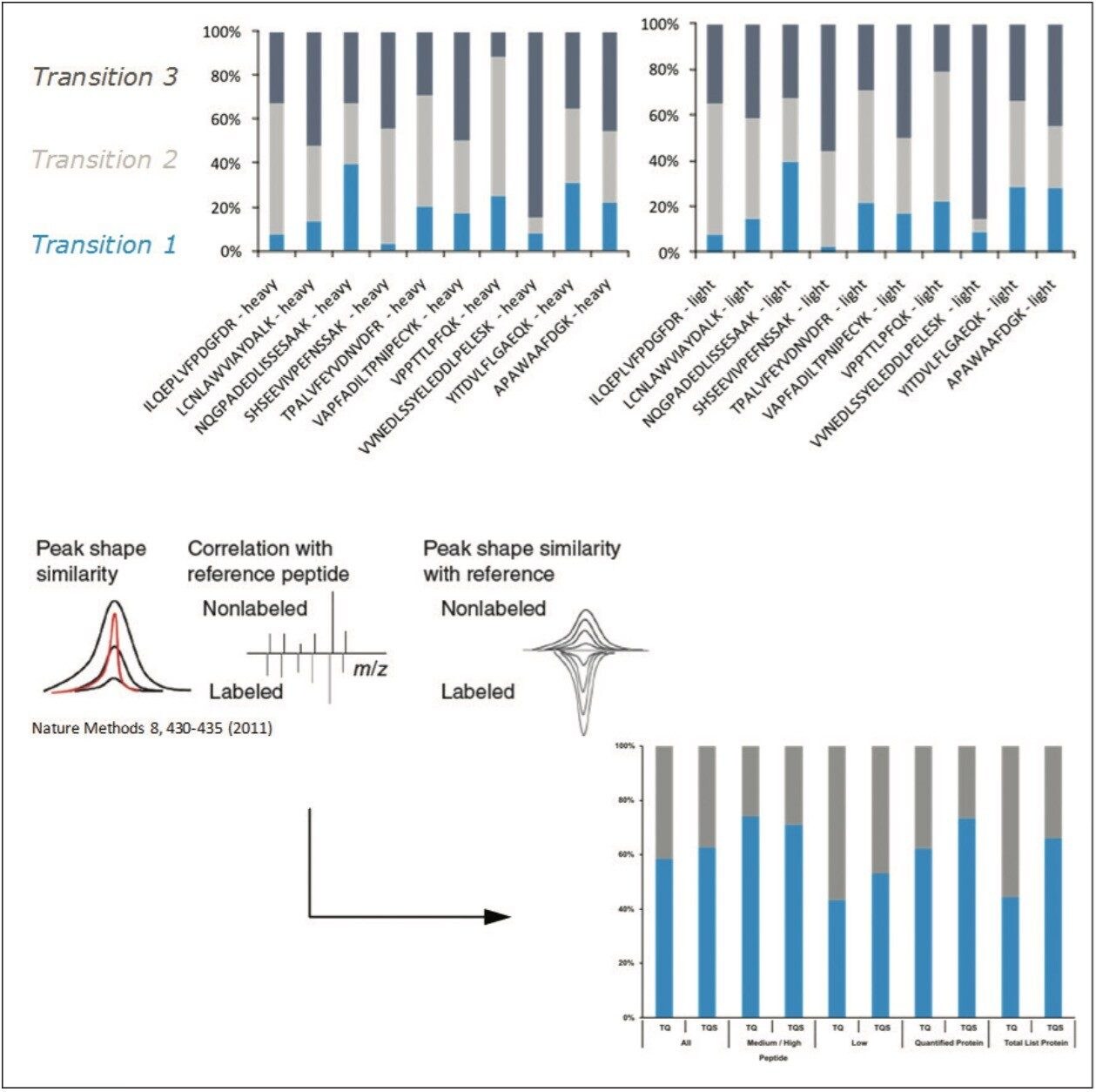
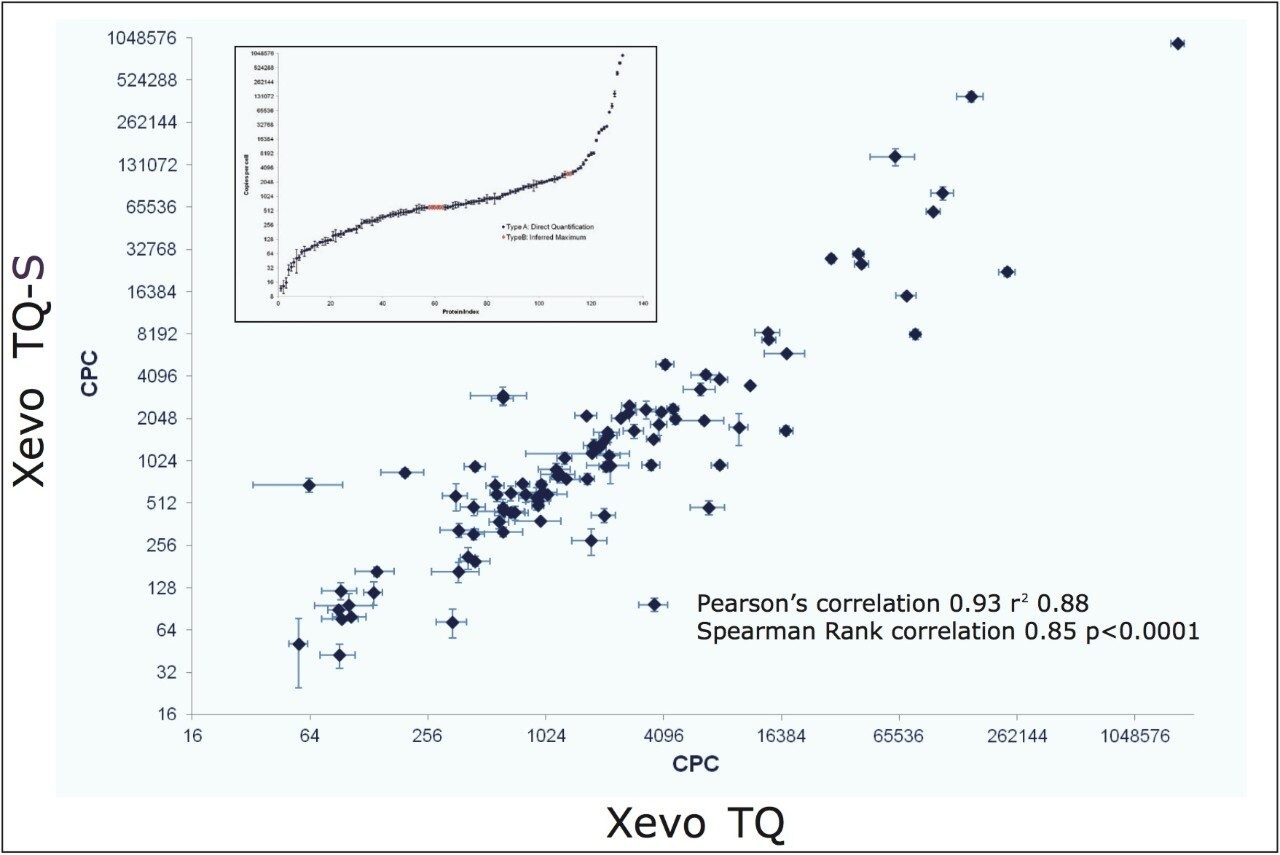
The design and quantification workflow of yeast kinases using QconCAT technology in high-throughput MRM mode is summarized in Figure 2. The top pane illustrates the design of a QconCAT internal standard protein and the bottom pane the workflow. The workflow comprised: i) digestion, ii) digestion performance check, iii) discovery LC-MS plus database searching, iv) creation spectral library, v) preliminary MRM quantitation, vi) transition validation, vii) experiment optimization, viii) serial dilutions, and ix) final quantitation.
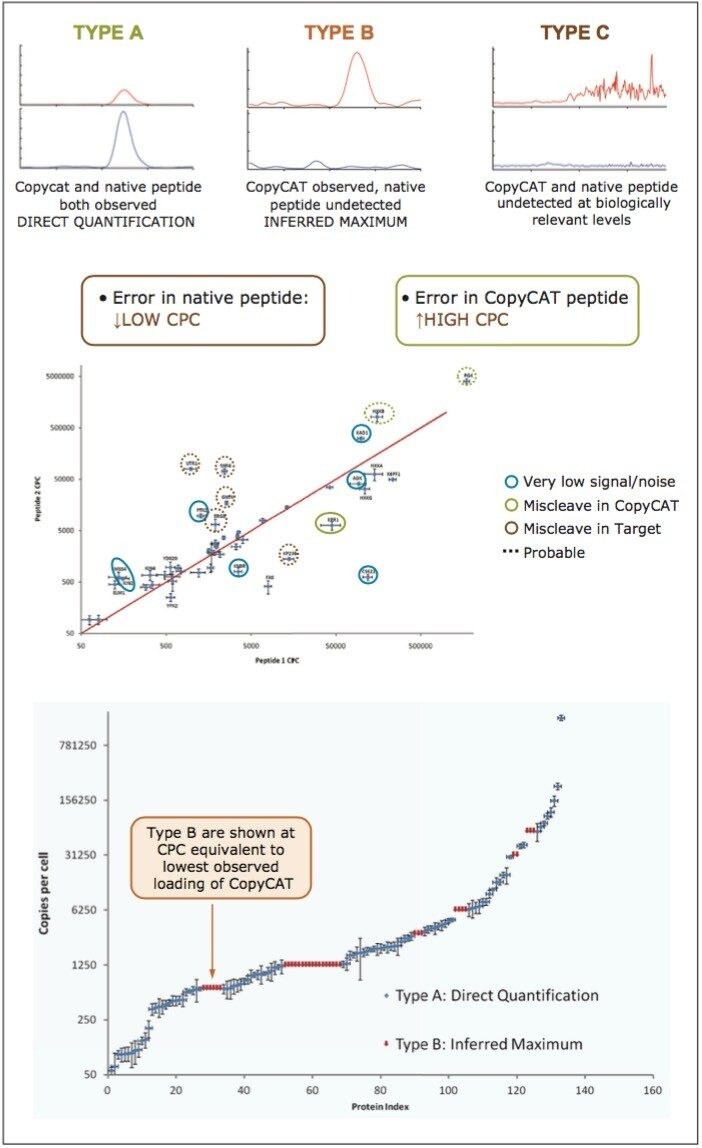
Peptides were qualified as either type A, B, or C, as illustrated in the top panel of Figure 4. A Type A identification infers unambiguous observation of both the QconCAT and native peptide. In the instance of Type B identifications, only the QconCAT peptide is observed and for Type C identifications both the QconCAT and native peptide were both not detected. The correlation between the selected peptides for Xevo TQ quantitation is illustrated in the middle pane of Figure 4. The majority of the peptides show good quantitative agreement; however, due to either low signal or missed tryptic cleavages, certain peptides cannot be used for quantitation, which are highlighted with blue, green, and red circles.