Manipulating Selectivity on HILIC
A method development scheme to analyze metformin and related substances presents a challenging task. Limitations regarding the low UV spectral absorbance of the analytes at 218 nm inhibit the use of typical MS-friendly buffers such as ammonium formate and ammonium acetate, since their UV cutoff approaches 230 nm. Reversed-phase LC is unsuccessful in retaining the analytes due to the polar basic characteristics of the compounds.
A method development scheme was employed to investigate two HILIC stationary phases: ACQUITY UPLC BEH HILIC and ACQUITY UPLC BEH Amide. Ionic strength, buffer cation selection, and temperature were determined as the remaining options to alter selectivity. Based on the poor retentivity and resolution of these compounds using the ACQUITY UPLC BEH HILIC stationary phase, the ACQUITY UPLC BEH Amide column was investigated.
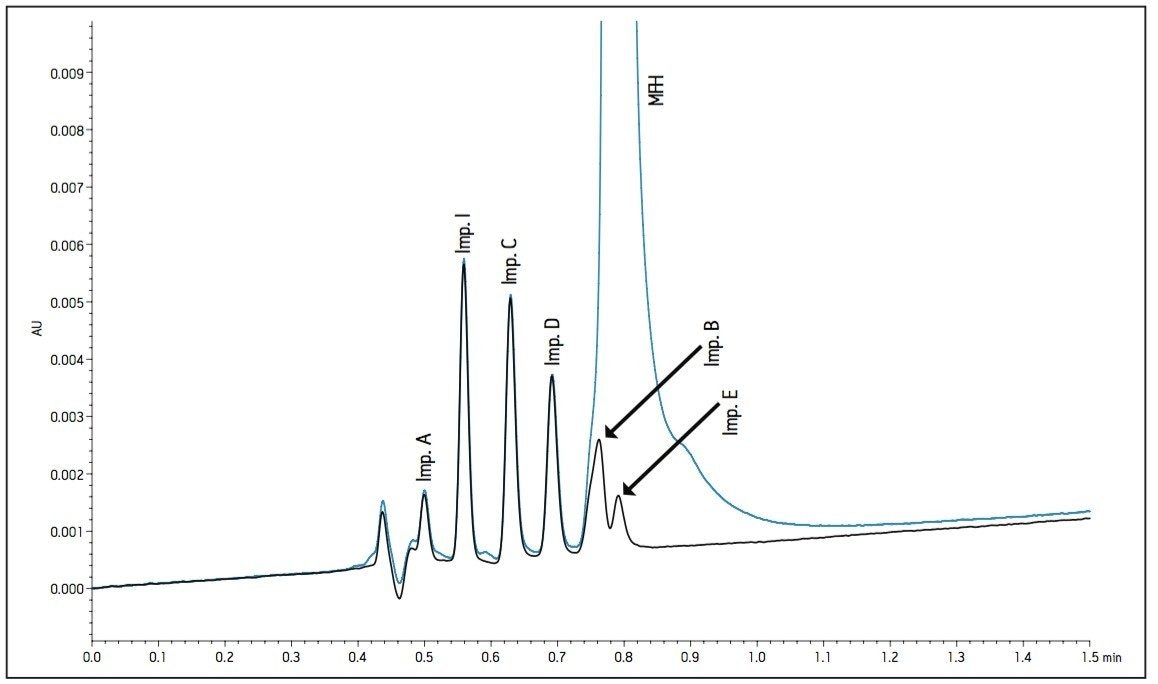
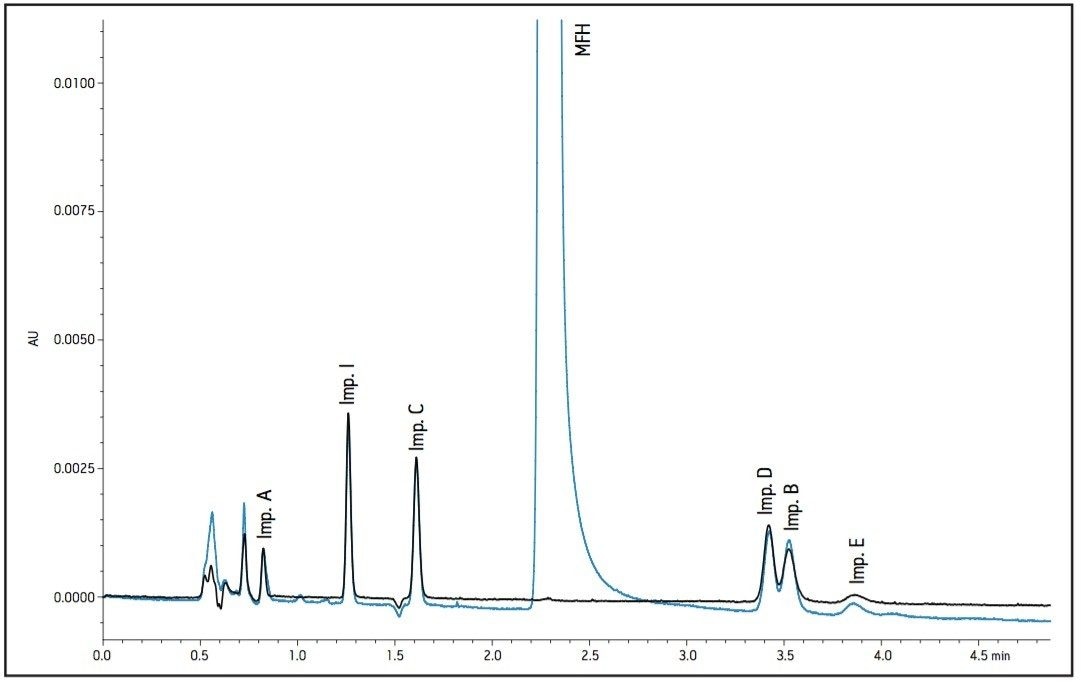
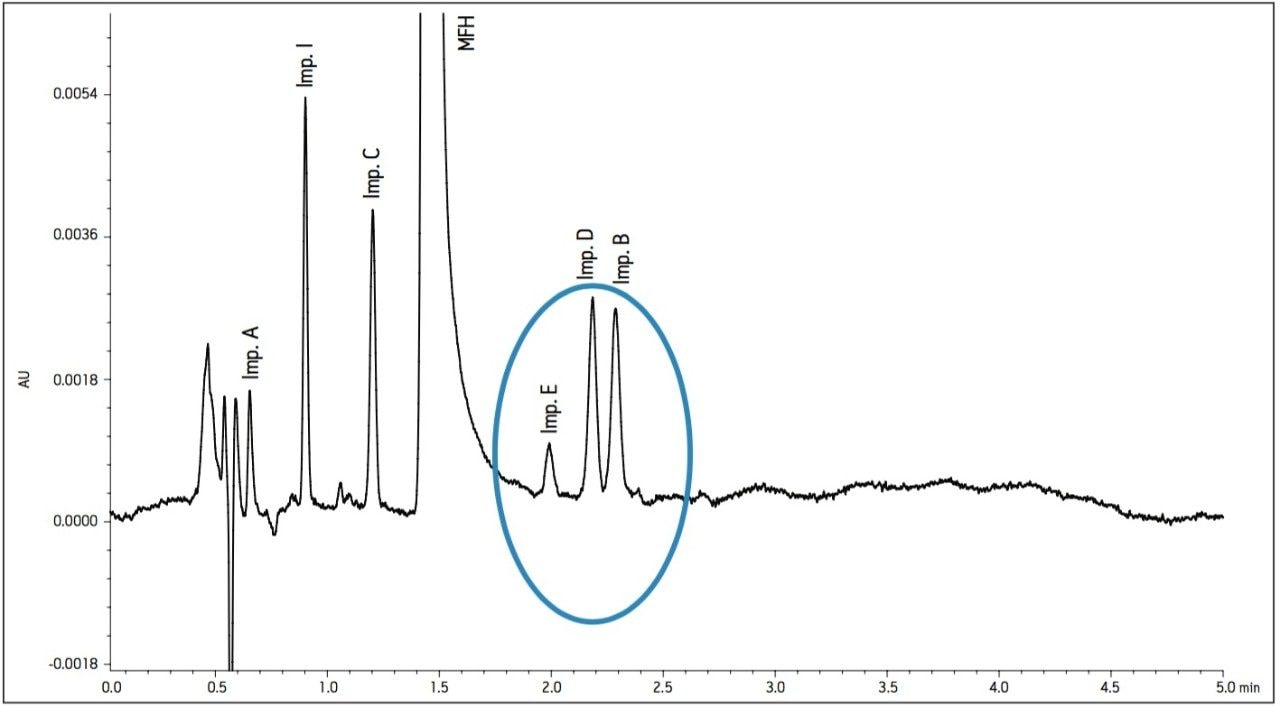
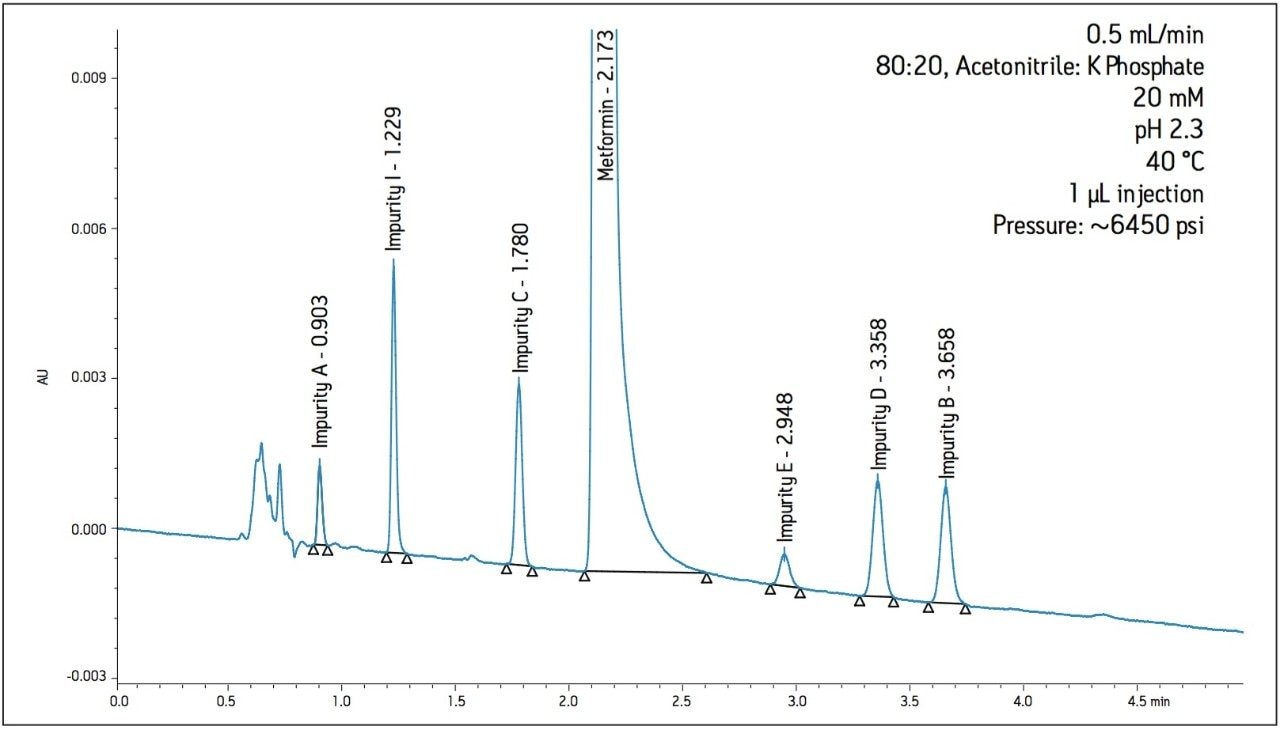
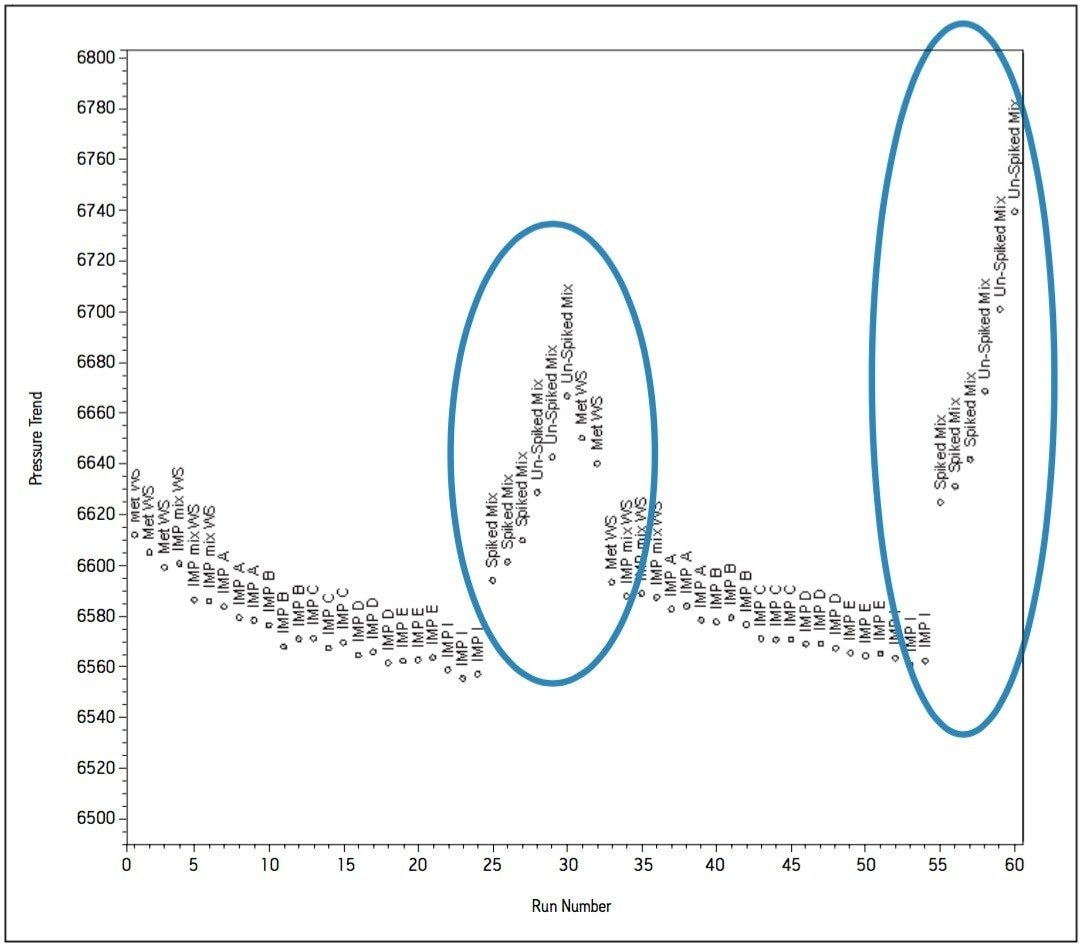
The working standard was injected onto the ACQUITY UPLC BEH Amide 2.1 x 100 mm, 1.7 μm column. The resulting chromatogram in Figure 2 resolved all compounds with the exception of a slight co-elution between Impurity B and Impurity D. Desired improvements in peak shape and sensitivity were seen for Impurity E. Due to the isocratic conditions, changes in flow rate and temperature were explored individually to improve on these critical impurity peaks of interest. An experiment utilizing higher mobilephase pH was explored but the results yielded little to no retention of many of the impurity peaks. The following relationships were observed during development of the separation on the amide column:
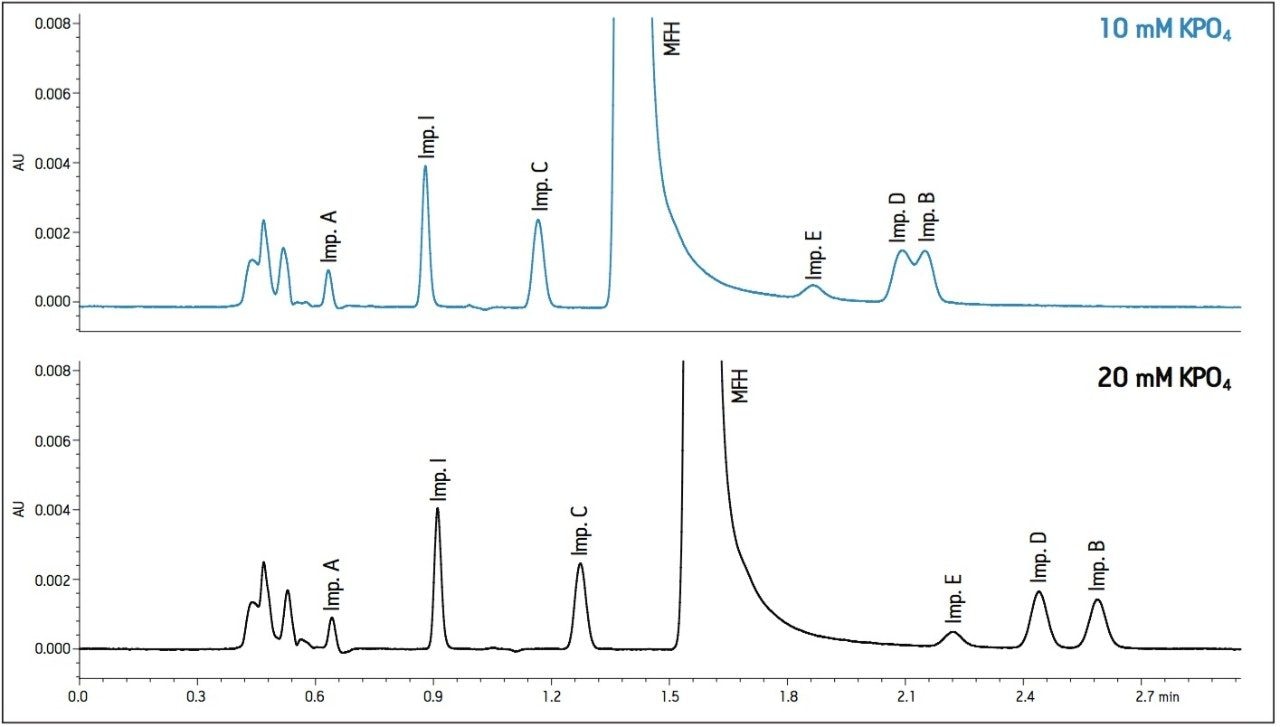
- Increases in organic content increased retention of Impurity D and B to co-elute with impurity E. A flow rate of 400 μL/min at 25 °C was determined to be optimal when using sodium phosphate as the aqueous mobile phase (Figure 2).
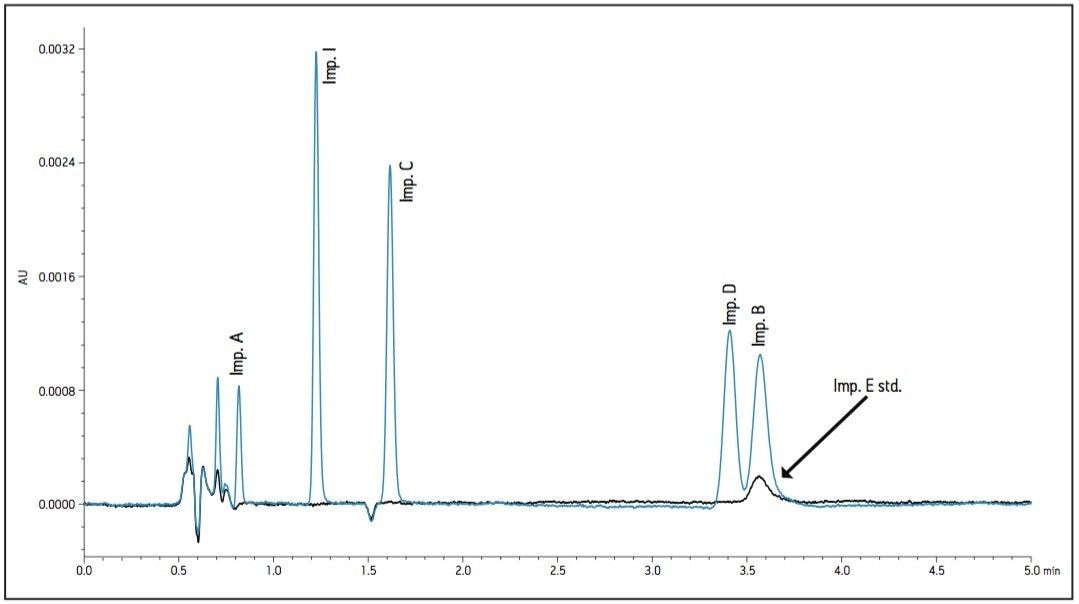
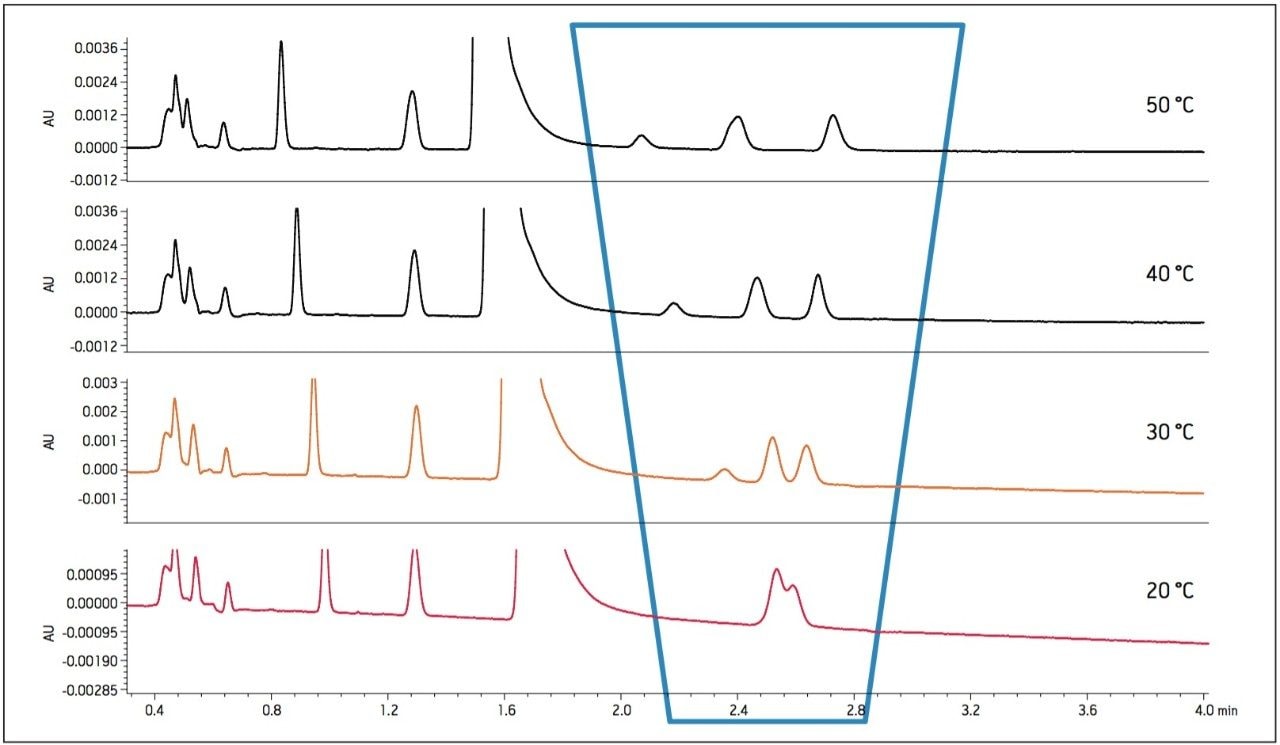
- As temperature increased, retention of impurity E decreased and co-eluted with impurity B (Figure 3).
- As flow rate increased, resolution of Impurity B, D, and E decreased.