The evaluation of pharmaceutical raw materials and finished products for impurities and degradation products is an essential part of the drug development and manufacturing testing process. It is a requirement that toxicological information be obtained on any drug-related impurity which is present at a concentration of greater than 0.1% of that of the active pharmaceutical ingredient (API). The impurities produced during the manufacturing process are often related to the solvents employed and raw material source, and so it is essential to compare the impurity profile when scaling from pilot to primary manufacturing, or when moving production from one location to another. It is also necessary to demonstrate the toxicological effects of any drug-related degradation products which are greater than this 0.1% API value.
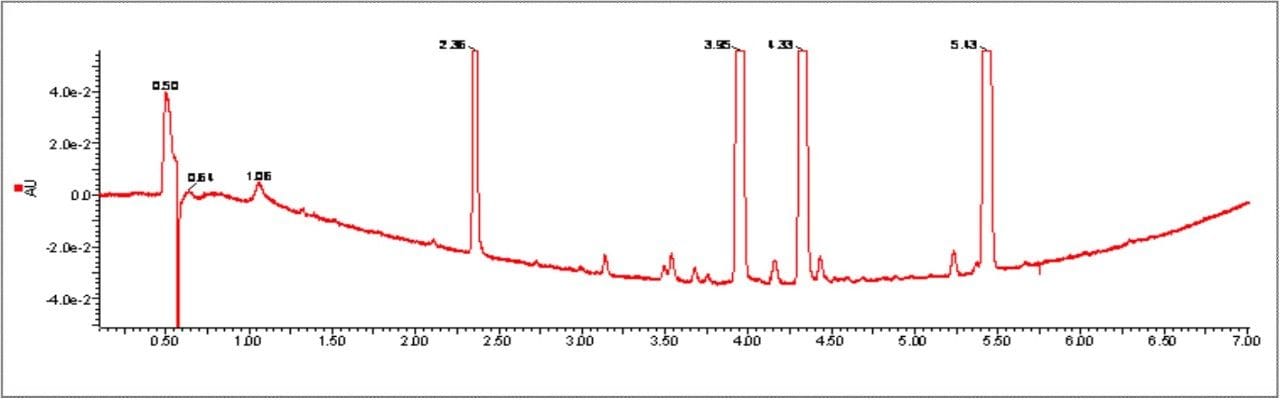
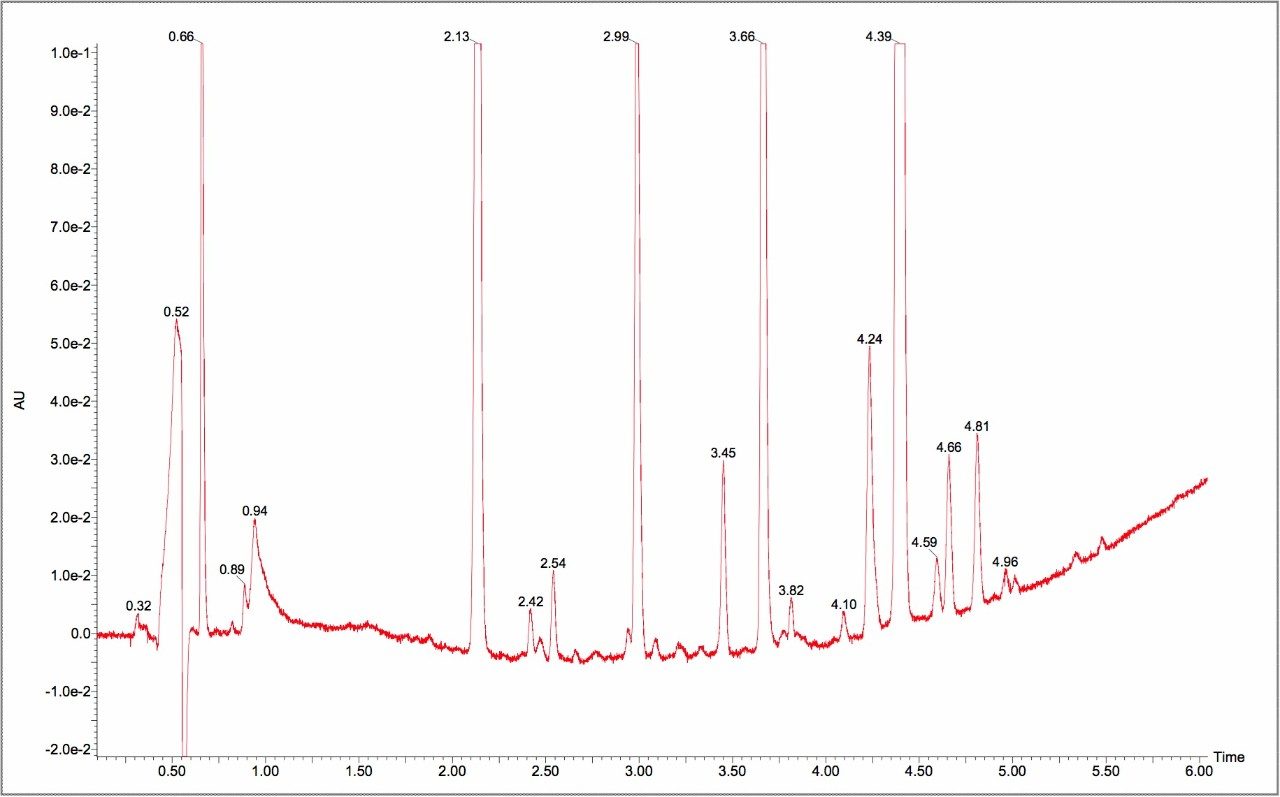
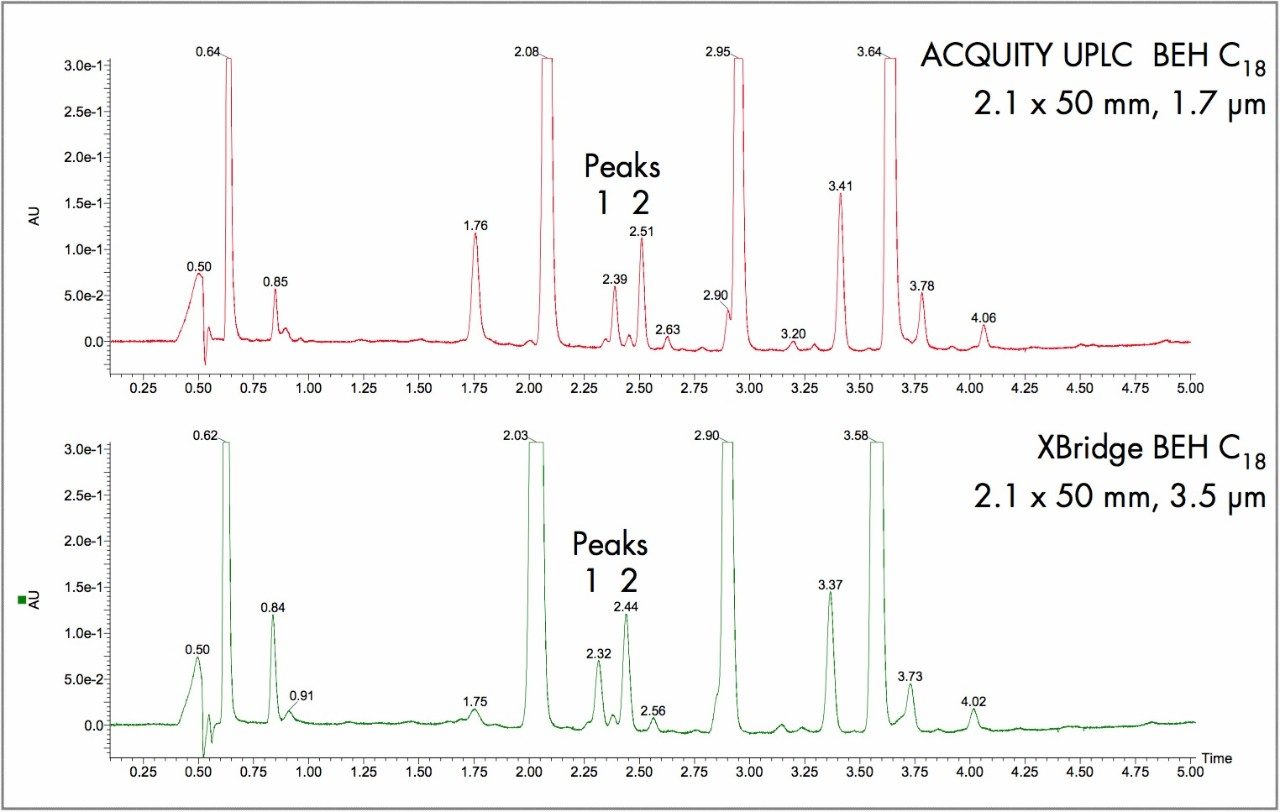
This impurity profiling is usually performed by HPLC with UV, PDA, or MS detection. As it is essential to detect and measure all of the impurities in the sample, it is necessary to have a high resolution separation process. This usually involves long analysis times resulting in low throughput.
As candidate pharmaceutical compounds become more potent and are dosed at lower and lower levels, there is a requirement for ever more sensitive assays to detect and measure impurities. This low throughput can become the rate-limiting step in product release testing or process evaluation. Any improvement in throughput and sensitivity would be of great benefit to the process of product release and identification of drug-related impurities. Since much of the process of impurity identification involves the coupling of liquid chromatography to sophisticated mass spectrometry equipment, any reduction in analysis time will result in a more efficient use of these significant investments.