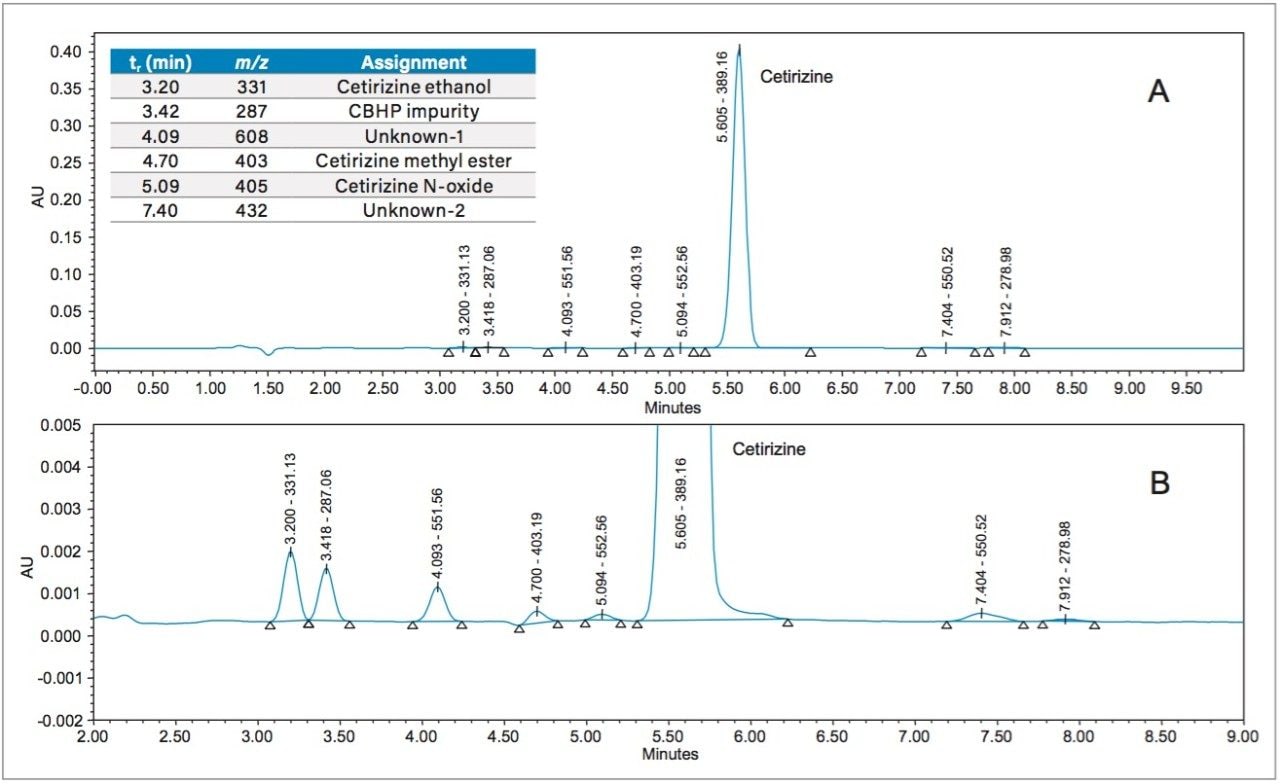
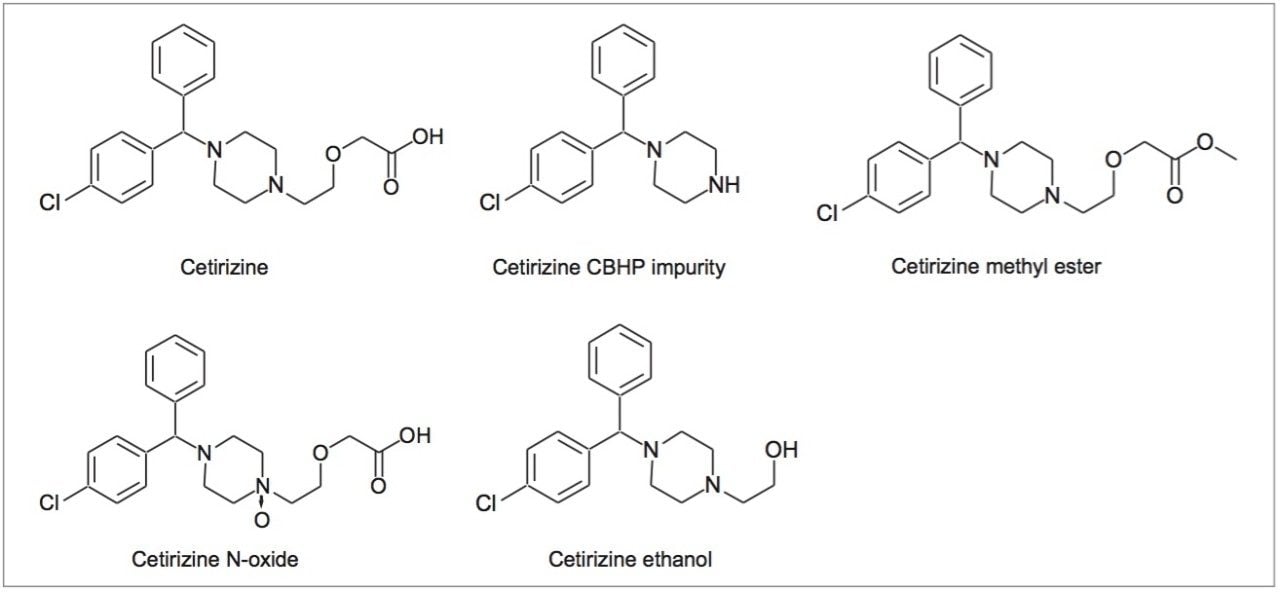
Cetirizine is a second-generation antihistamine that is used in the treatment of hay fever, urticaria, angioedema, and allergies. The USP method for organic impurities in cetirizine hydrochloride tablets specifies the use of a 4.0 x 250 mm, 5 μm L3 column (porous silica particles, 1.5 μm–10 μm diameter, or a monolithic rod).1 Permissible alterations are given in USP General Chapter <621>, according to which, for isocratic methods, particle size (dp) and/or column length (L) can be changed as long as the ratio of column length to particle size (L/dp) remains constant or within the range -25% to +50% of the original column specified. Alternatively, other combinations of L and dp may be employed provided the number of theoretical plates (N) is within the range -25% to +50% of the original column specified in the method.2
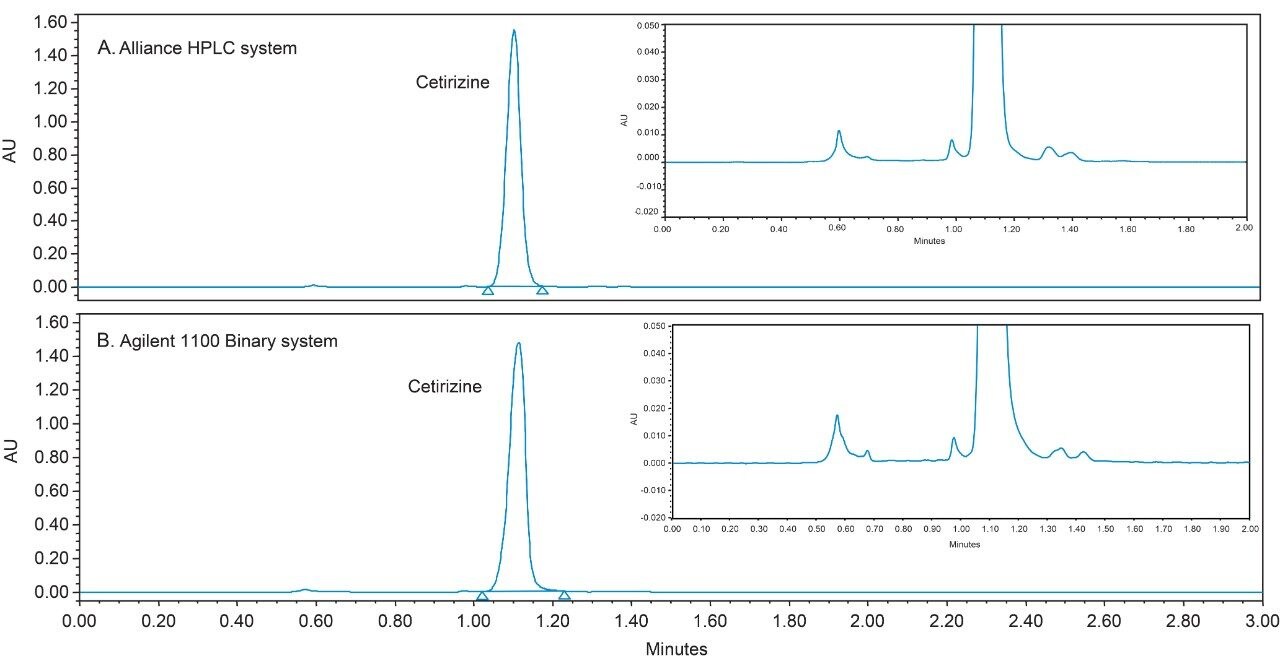
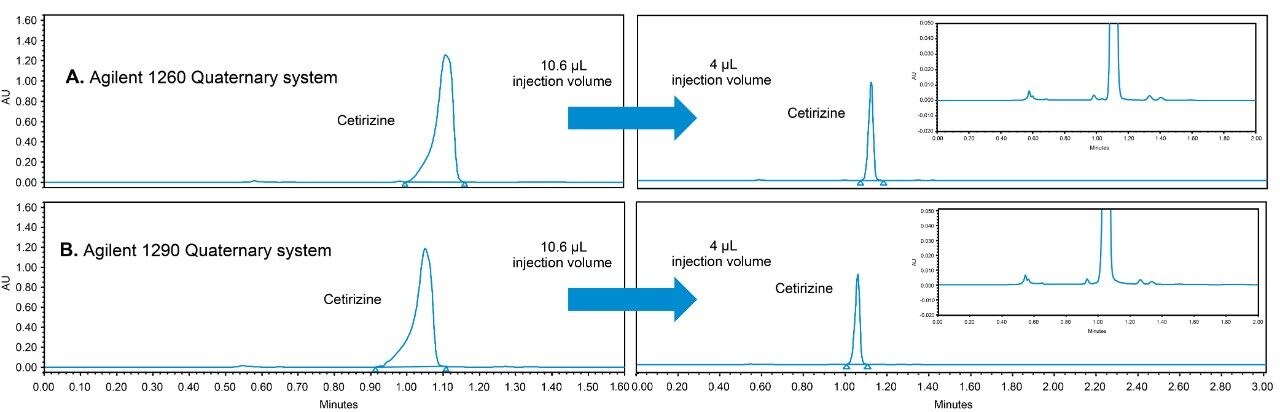
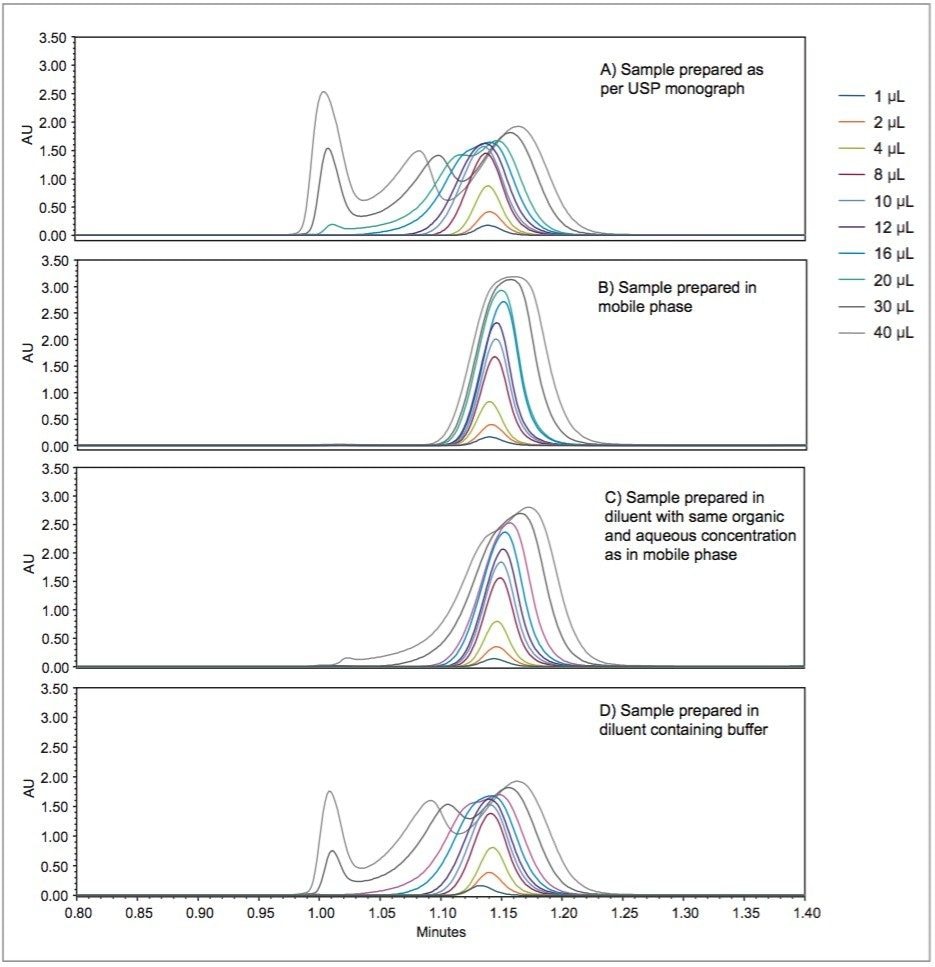
Hence, an existing USP method can be modernized by using newer column chemistries without the need to revalidate the method. Columns containing smaller particles substantially reduce analysis times without compromising the quality of the data, when the flow rate is scaled accordingly. The organic impurity method for cetirizine hydrochloride tablets has been updated by using an XBridge HILIC XP, 2.5 μm, 4.6 x 100 mm Column (p/n: 186006087).3 The USP allowable changes reduced the analysis time by a factor of 5 (from 15 minutes to 3 minutes) and improved the peak shape for cetirizine HCl. The peak shape improvement was due to injecting a scaled, smaller injection volume, which mitigated much of the peak distortion caused by the sample diluent. Further evidence of sample diluent induced peak distortion will be presented in this work.
Also Run On:
Agilent 1100 Binary System
Agilent 1260 Infinity Quaternary System
Agilent 1290 Infinity Quaternary System
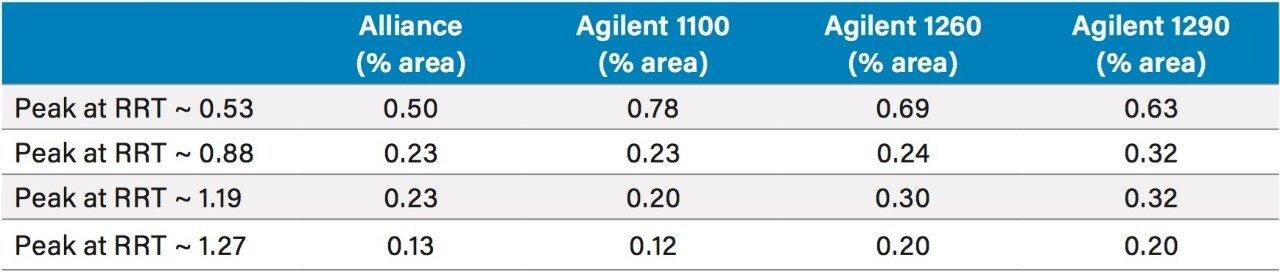
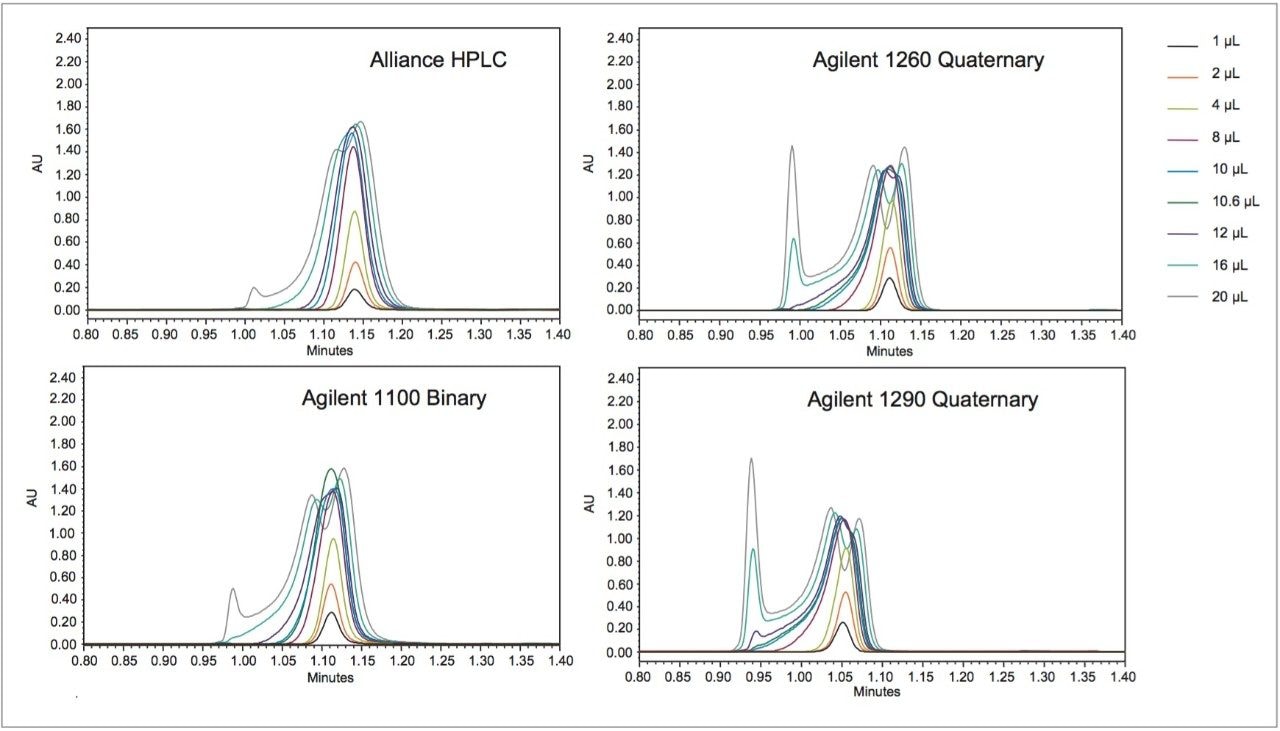
USP methods are validated and are generally accepted to work on any properly functioning LC system. However, few examples of successful HILIC transfers between systems from different vendors have been shown. The same HILIC method and column were tested on four different LC systems, from Waters (Alliance HPLC System) and Agilent (1100 Binary HPLC System, 1260 Infinity Quaternary UHPLC System and 1290 Infinity Quaternary UHPLC System), each controlled by Empower CDS. The particular method used required some investigation, in which a key obstacle was overcome to achieve undistorted peaks.
The method was then modified using a MS-compatible mobile phase to demonstrate the feasibility of further modernizing the method.