Paralytic shellfish toxins (PST) are a naturally-occurring family of marine biotoxins, termed saxitoxins, produced by certain species of algae and bacteria, and reported globally. These algae are periodically found at high cell densities in the sea, during which they can accumulate in bivalve shellfish such as mussels, oysters, and clams.1 Tetrodotoxin (TTX) is another biotoxin, thought to be produced by marine bacteria, which has been found to accumulate in the tissues of shellfish.2 These toxins are potent neurotoxins, so may give rise to paralytic shellfish poisoning (PSP) in human consumers of contaminated products, resulting in the need for routine official control testing and end product testing of bivalve mollusks. In Europe, Regulation (EC) No. 854/2004, which has become part of Regulation (EC) 2017/625 as part of the revision of official control provision, requires a monitoring program of classified shellfish production areas to be established as part of the competent authority’s official controls to check for the possible presence of marine biotoxins in the shellfish flesh.3 All food business operators are required to ensure that they only place on the market food that is both safe and compliant with relevant requirements.
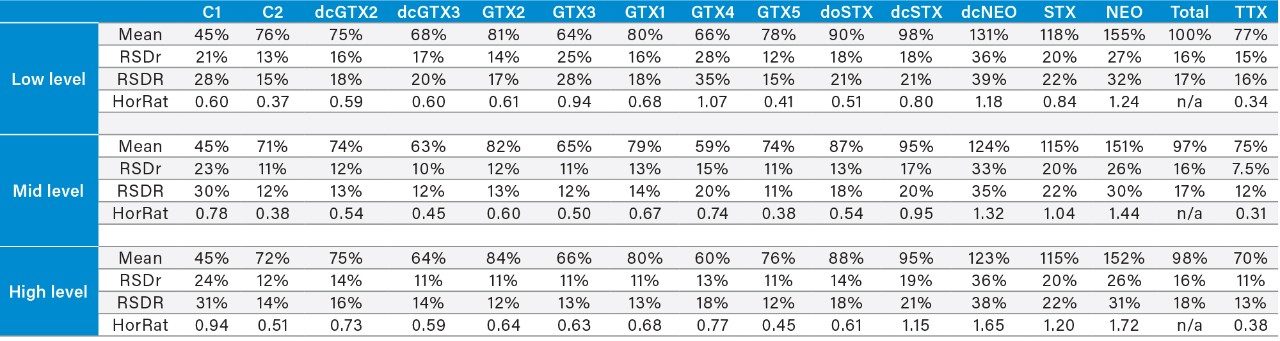
Both commercial and regulatory testing laboratories have interest in the need for a method that is simple, highly sensitive in relation to limits set in many countries, quick to use, and provides accurate, precise, and reproducible results. Most regulations around the world set maximum permitted levels (MPL) for PSP toxins as a group, typically 800 μg STX eq/kg of shellfish meat. Regulatory limits for marine biotoxins in shellfish in Europe are laid down in Regulation (EC) No. 853/2004.4
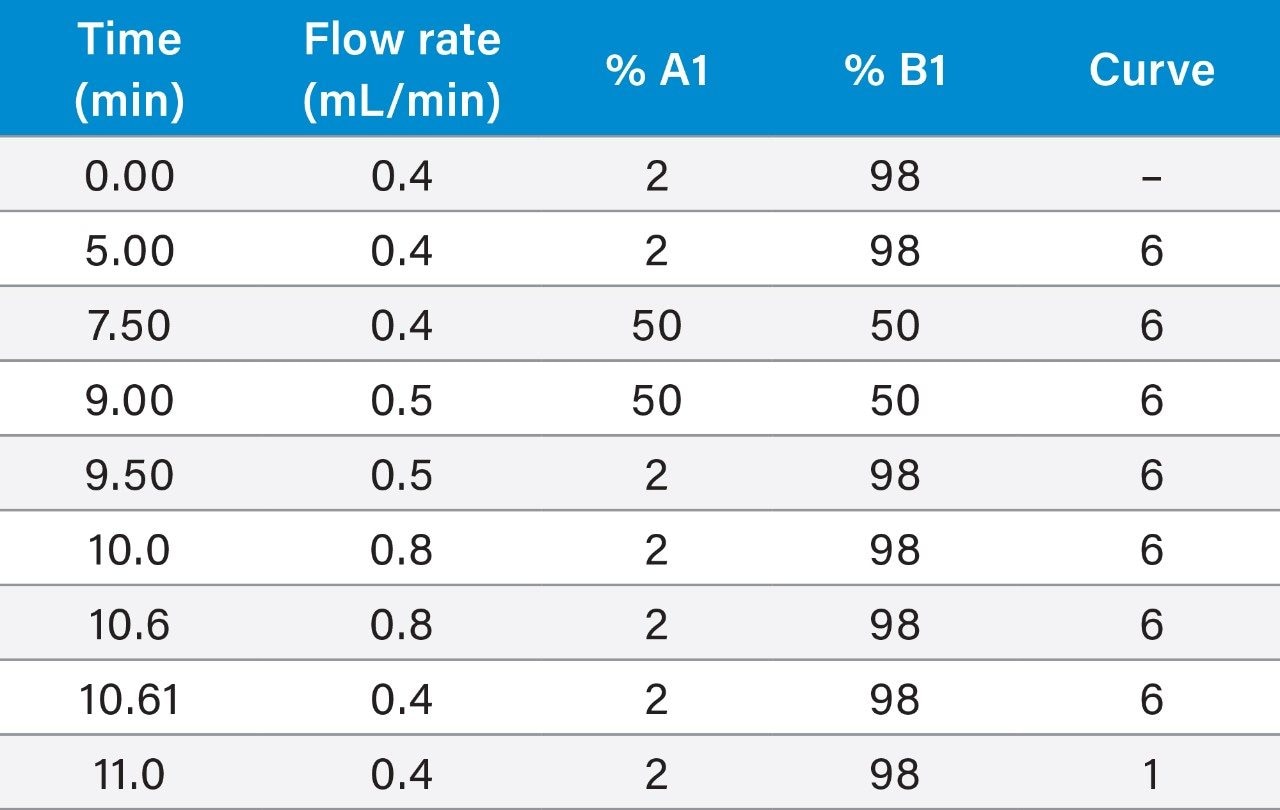
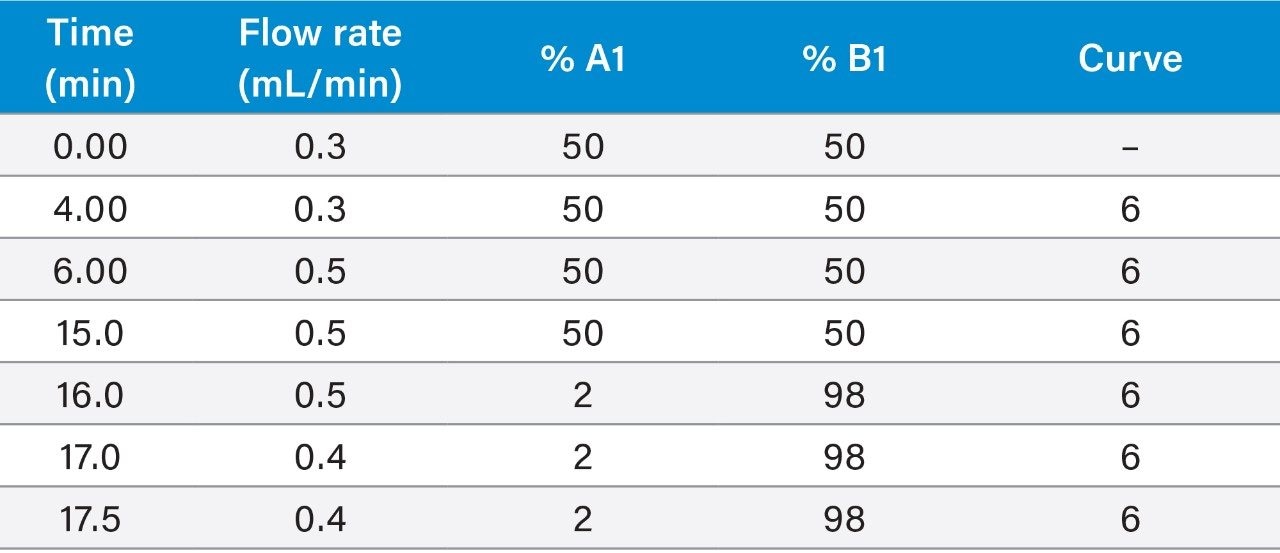
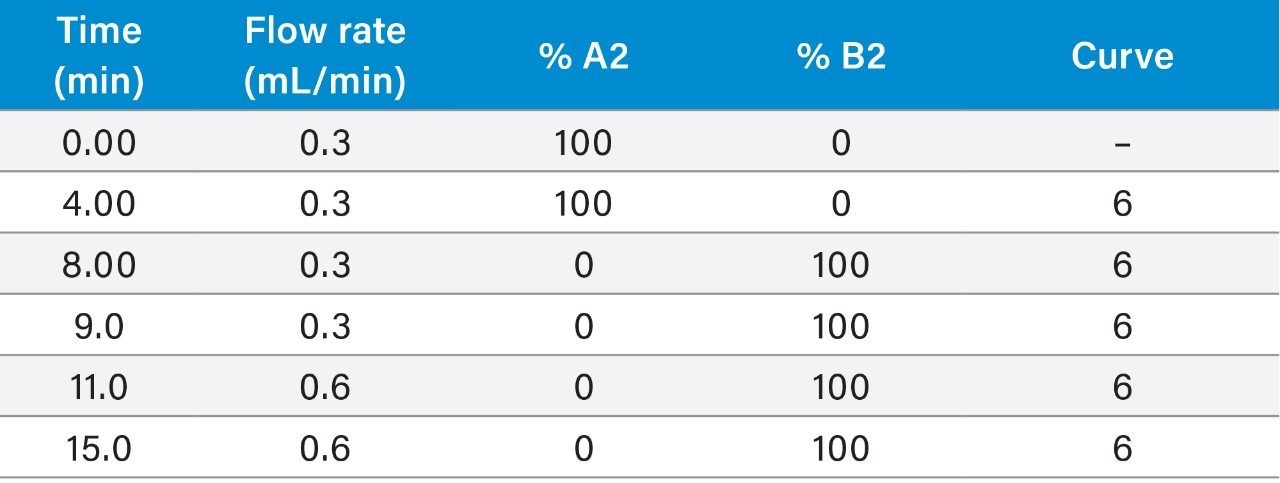
Within the European Union (EU), the official reference method for PST is AOAC OMA 2005.06 based on a pre-column oxidation liquid chromatography (LC) with fluorescence detection.5 While this provides an excellent level of regulatory control, the method is complex and time-consuming, requiring multiple clean-ups, derivatizations and analytical runs per sample, so a rapid one-shot method of analysis for PST is desirable.
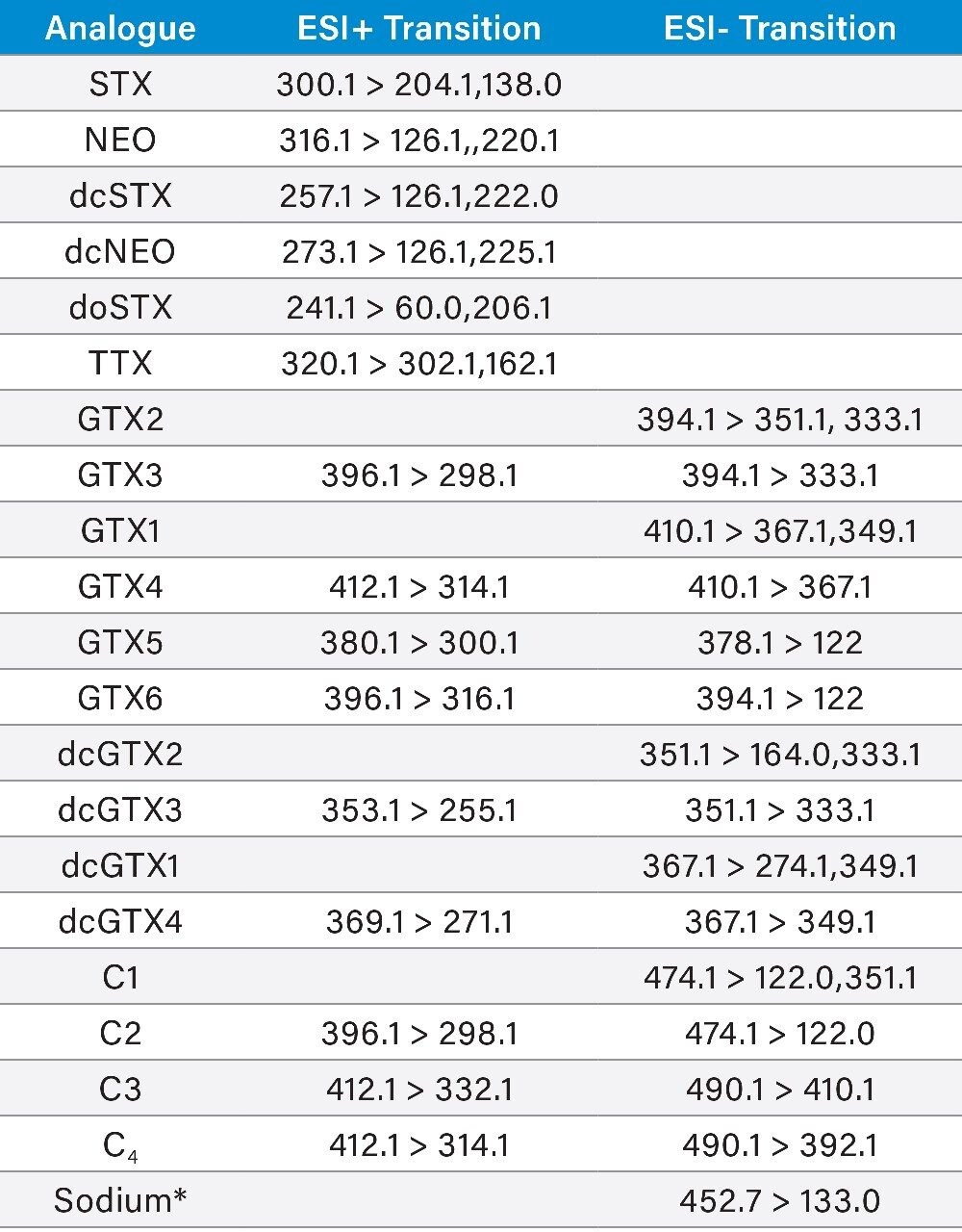
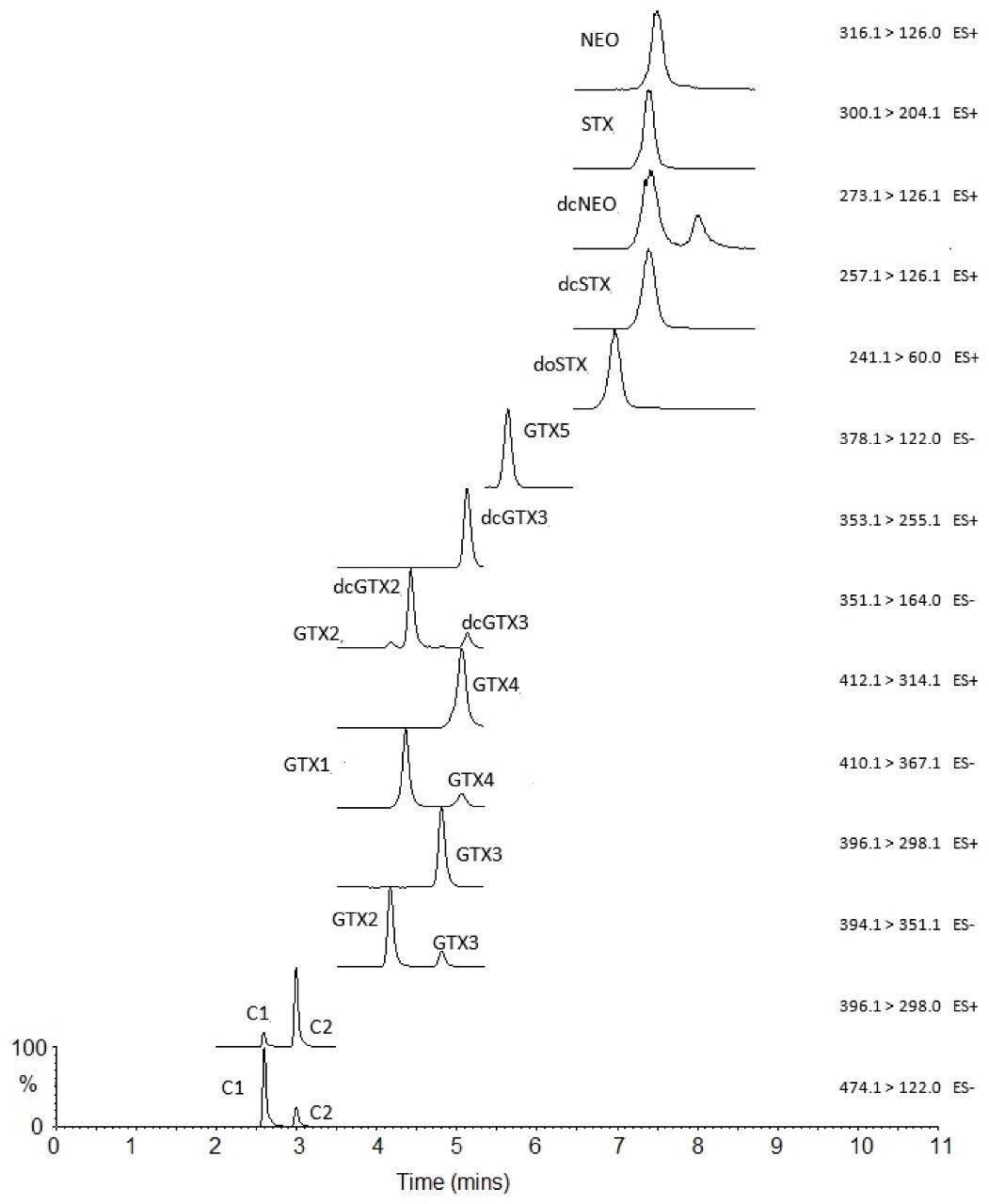
The LC-FLD method is also unable to detect TTX, which has been found in shellfish throughout Europe in recent years. There is consequently great interest in the application of a recently developed method involving hydrophilic interaction liquid chromatography with tandem mass spectrometric detection (HILIC-MS/MS).6 HILIC-MS/MS utilizing ultra-performance liquid chromatography (UPLC) has been validated for PST7 and TTX8 and is currently undergoing further validation through collaborative study.
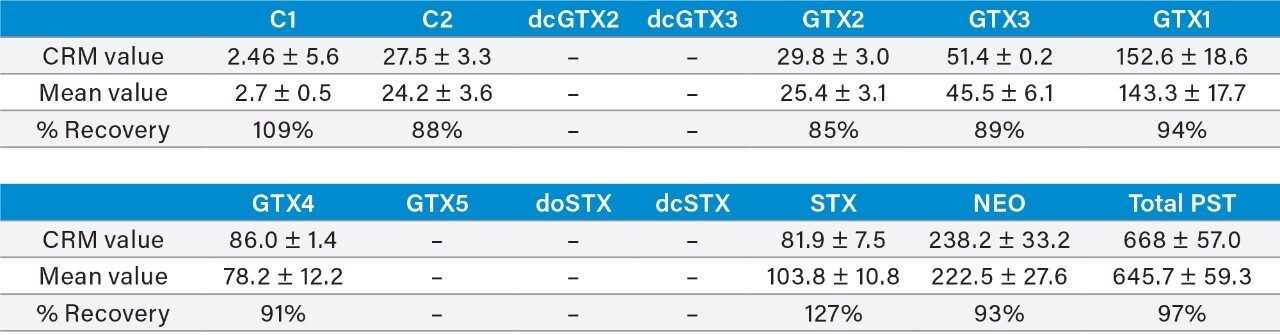
This application note reports the results of the rapid, single-dispersive extraction, graphitized carbon-SPE HILIC-MS/MS method, validated for the determination of PSTs and TTX in shellfish.